

The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country
- PMID: 32752159
- PMCID: PMC7466227
- DOI: 10.3390/foods9081030
The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country
Abstract
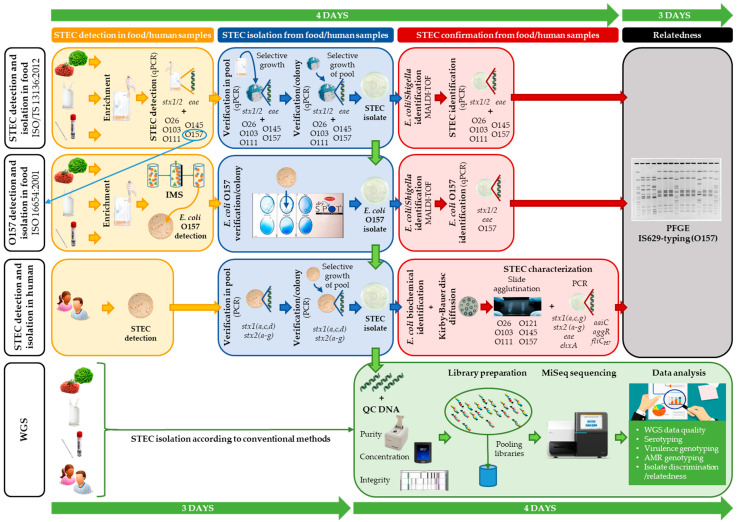
Gradually, conventional methods for foodborne pathogen typing are replaced by whole genome sequencing (WGS). Despite studies describing the overall benefits, National Reference Laboratories of smaller countries often show slower uptake of WGS, mainly because of significant investments required to generate and analyze data of a limited amount of samples. To facilitate this process and incite policy makers to support its implementation, a Shiga toxin-producing Escherichia coli (STEC) O157:H7 (stx1+, stx2+, eae+) outbreak (2012) and a STEC O157:H7 (stx2+, eae+) outbreak (2013) were retrospectively analyzed using WGS and compared with their conventional investigations. The corresponding results were obtained, with WGS delivering even more information, e.g., on virulence and antimicrobial resistance genotypes. Besides a universal, all-in-one workflow with less hands-on-time (five versus seven actual working days for WGS versus conventional), WGS-based cgMLST-typing demonstrated increased resolution. This enabled an accurate cluster definition, which remained unsolved for the 2013 outbreak, partly due to scarce epidemiological linking with the suspect source. Moreover, it allowed detecting two and one earlier circulating STEC O157:H7 (stx1+, stx2+, eae+) and STEC O157:H7 (stx2+, eae+) strains as closely related to the 2012 and 2013 outbreaks, respectively, which might have further directed epidemiological investigation initially. Although some bottlenecks concerning centralized data-sharing, sampling strategies, and perceived costs should be considered, we delivered a proof-of-concept that even in smaller countries, WGS offers benefits for outbreak investigation, if a sufficient budget is available to ensure its implementation in surveillance. Indeed, applying a database with background isolates is critical in interpreting isolate relationships to outbreaks, and leveraging the true benefit of WGS in outbreak investigation and/or prevention.
Keywords: STEC; Shiga toxin-producing Escherichia coli; WGS; food safety; foodborne outbreak investigation; surveillance; whole genome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- World Health Organization (WHO) Foodborne Disease Outbreaks: Guidelines for Investigation and Control. World Health Organization; Geneva, Switzerland: 2008.
-
- Baker C.A., Rubinelli P.M., Park S.H., Carbonero F., Ricke S.C. Shiga Toxin-Producing Escherichia coli in Food: Incidence, Ecology, and Detection Strategies. Food Control. 2016;59:407–419. doi: 10.1016/j.foodcont.2015.06.011. - DOI
-
- Ooka T., Ogura Y., Asadulghani M., Ohnishi M., Nakayama K., Terajima J., Watanabe H., Hayashi T. Inference of the Impact of Insertion Sequence (IS) Elements on Bacterial Genome Diversification through Analysis of Small-Size Structural Polymorphisms in Escherichia coli O157 Genomes. Genome Res. 2009;19:1809–1816. doi: 10.1101/gr.089615.108. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
