

Complex landscape of alternative splicing in myeloid neoplasms
- PMID: 32753690
- PMCID: PMC8101081
- DOI: 10.1038/s41375-020-1002-y
Complex landscape of alternative splicing in myeloid neoplasms
Erratum in
-
Correction: complex landscape of alternative splicing in myeloid neoplasms.Leukemia. 2021 Apr;35(4):1226. doi: 10.1038/s41375-021-01197-2. Leukemia. 2021. PMID: 33714977 Free PMC article. No abstract available.
Abstract
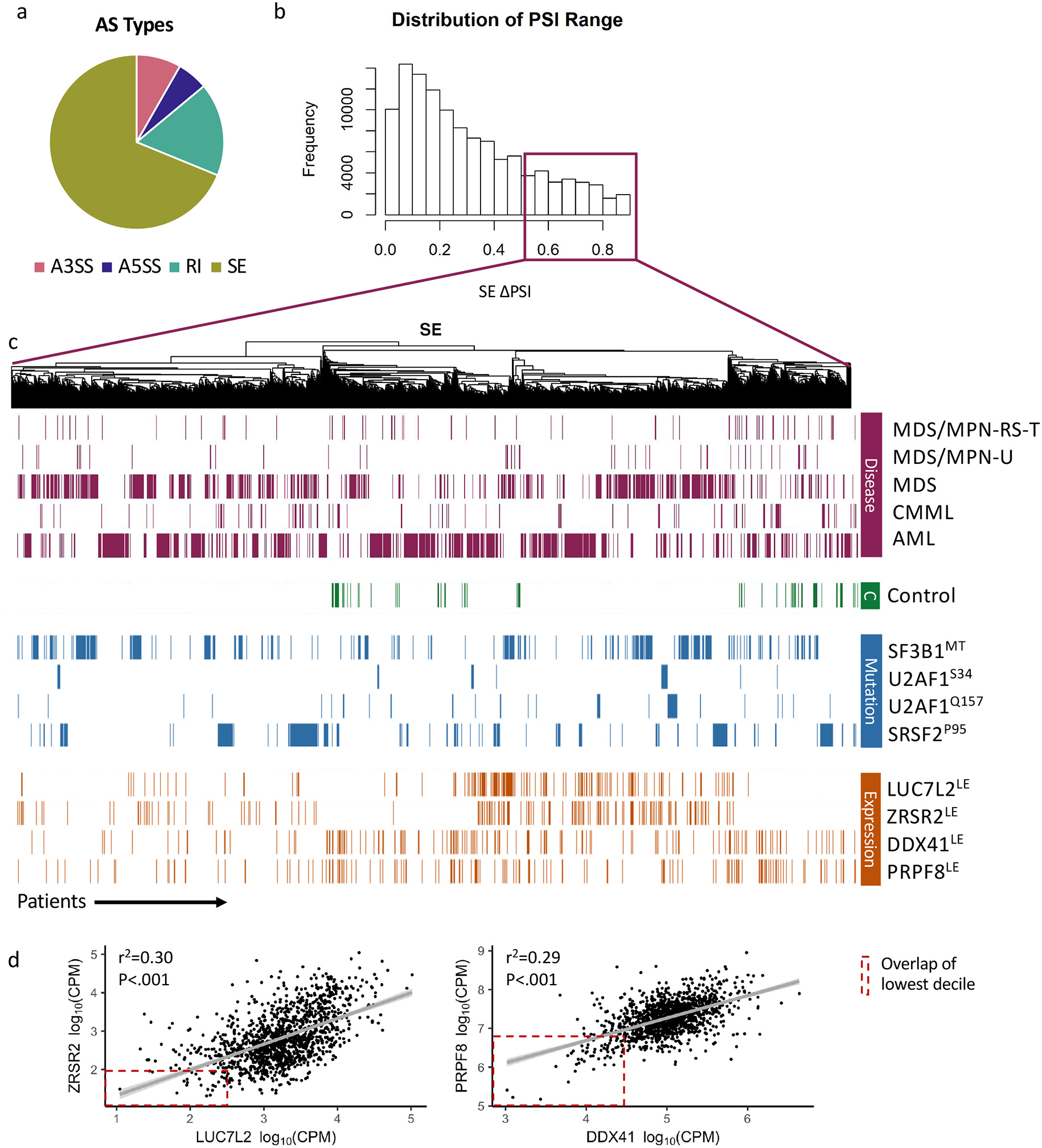
Myeloid neoplasms are characterized by frequent mutations in at least seven components of the spliceosome that have distinct roles in the process of pre-mRNA splicing. Hotspot mutations in SF3B1, SRSF2, U2AF1 and loss of function mutations in ZRSR2 have revealed widely different aberrant splicing signatures with little overlap. However, previous studies lacked the power necessary to identify commonly mis-spliced transcripts in heterogeneous patient cohorts. By performing RNA-Seq on bone marrow samples from 1258 myeloid neoplasm patients and 63 healthy bone marrow donors, we identified transcripts frequently mis-spliced by mutated splicing factors (SF), rare SF mutations with common alternative splicing (AS) signatures, and SF-dependent neojunctions. We characterized 17,300 dysregulated AS events using a pipeline designed to predict the impact of mis-splicing on protein function. Meta-splicing analysis revealed a pattern of reduced levels of retained introns among disease samples that was exacerbated in patients with splicing factor mutations. These introns share characteristics with "detained introns," a class of introns that have been shown to promote differentiation by detaining pro-proliferative transcripts in the nucleus. In this study, we have functionally characterized 17,300 targets of mis-splicing by the SF mutations, identifying a common pathway by which AS may promote maintenance of a proliferative state.
Conflict of interest statement
Competing Interests
T.H., C.H. and W.K. serve as the management board of the MLL Munich Leukemia Laboratory. N.N, S.T., S.H, M.M., W.W., and C.B. are employed by the MLL Munich Leukemia Laboratory. The MLL offers diagnostic services for leukaemias and lymphomas, including cytomorphology, cytochemistry, immunophenotyping, cytogenetics, FISH, and a broad spectrum of molecular assays. In addition, MLL runs several research studies based on a combination of methods for routine use and also including whole genome sequencing and whole transcriptome sequencing.
Figures





References
-
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478 (7367):64–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
