

Computational methods for exploring protein conformations
- PMID: 32756904
- PMCID: PMC7458412
- DOI: 10.1042/BST20200193
Computational methods for exploring protein conformations
Abstract
Proteins are dynamic molecules that can transition between a potentially wide range of structures comprising their conformational ensemble. The nature of these conformations and their relative probabilities are described by a high-dimensional free energy landscape. While computer simulation techniques such as molecular dynamics simulations allow characterisation of the metastable conformational states and the transitions between them, and thus free energy landscapes, to be characterised, the barriers between states can be high, precluding efficient sampling without substantial computational resources. Over the past decades, a dizzying array of methods have emerged for enhancing conformational sampling, and for projecting the free energy landscape onto a reduced set of dimensions that allow conformational states to be distinguished, known as collective variables (CVs), along which sampling may be directed. Here, a brief description of what biomolecular simulation entails is followed by a more detailed exposition of the nature of CVs and methods for determining these, and, lastly, an overview of the myriad different approaches for enhancing conformational sampling, most of which rely upon CVs, including new advances in both CV determination and conformational sampling due to machine learning.
Keywords: collective variables; conformational ensemble; enhanced sampling; machine learning; molecular dynamics; proteins.
© 2020 The Author(s).
Conflict of interest statement
The author declares that there are no competing interests associated with this manuscript.
Figures
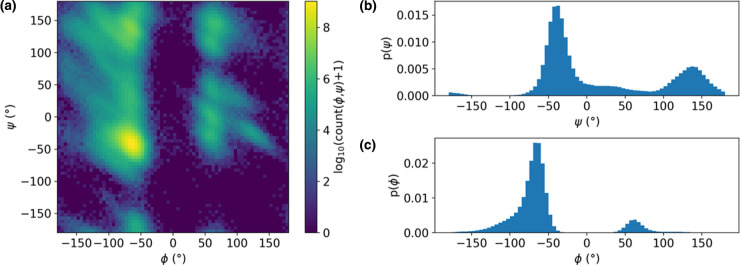
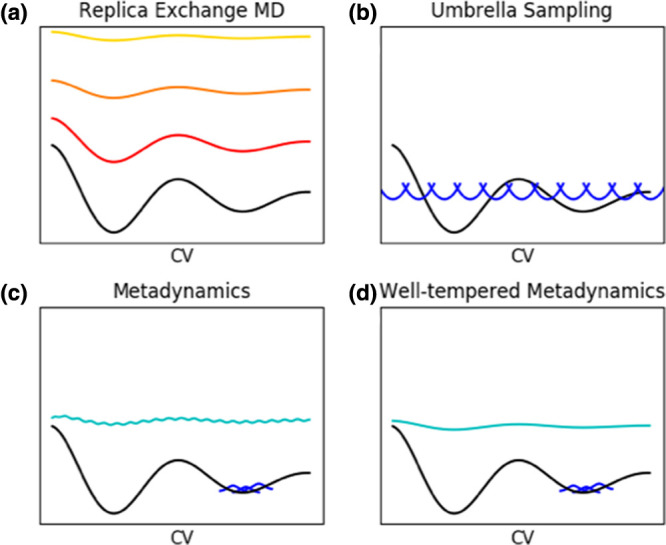
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous