

Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis
- PMID: 32759504
- PMCID: PMC7598040
- DOI: 10.1172/JCI141374
Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis
Abstract
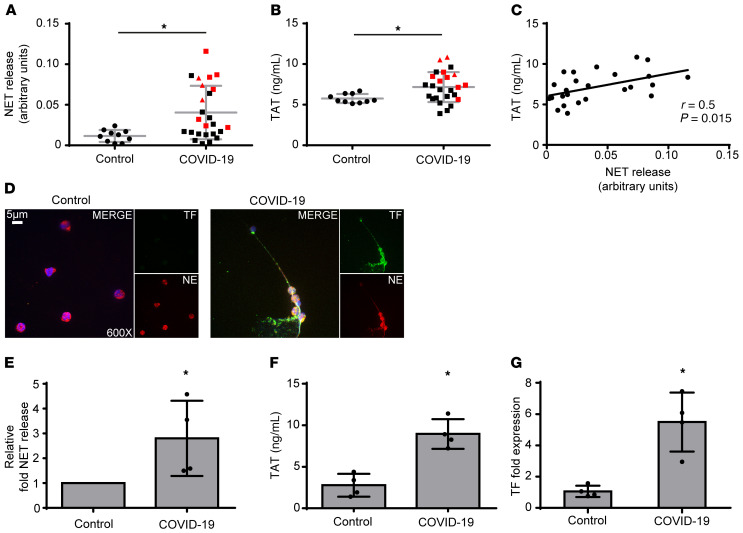
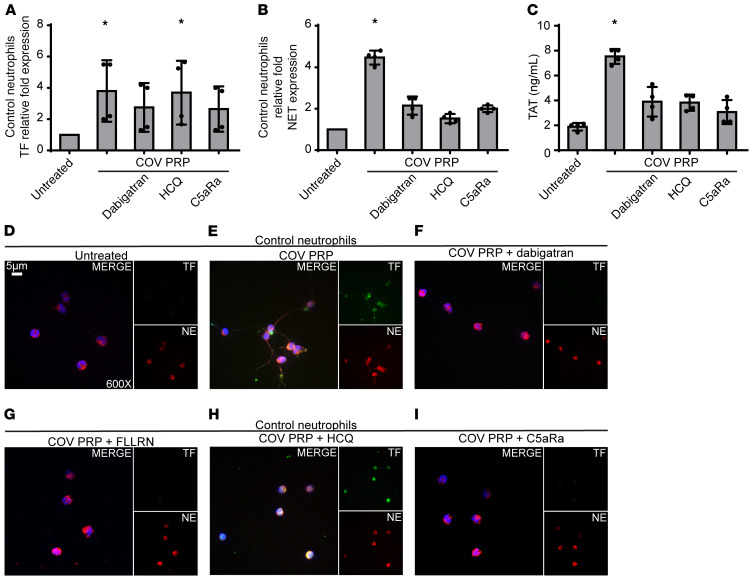
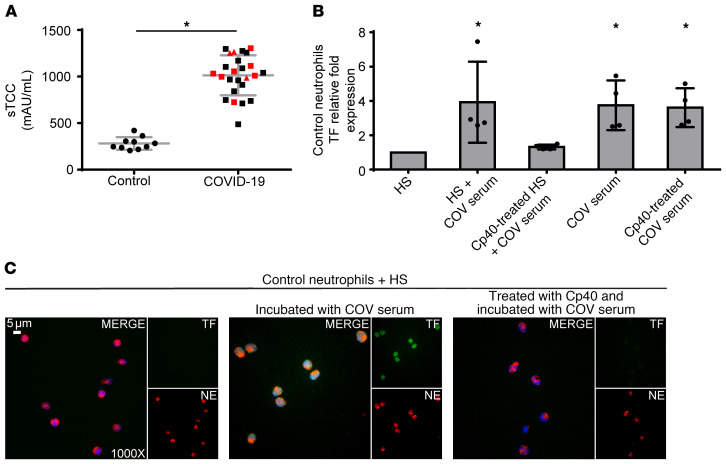
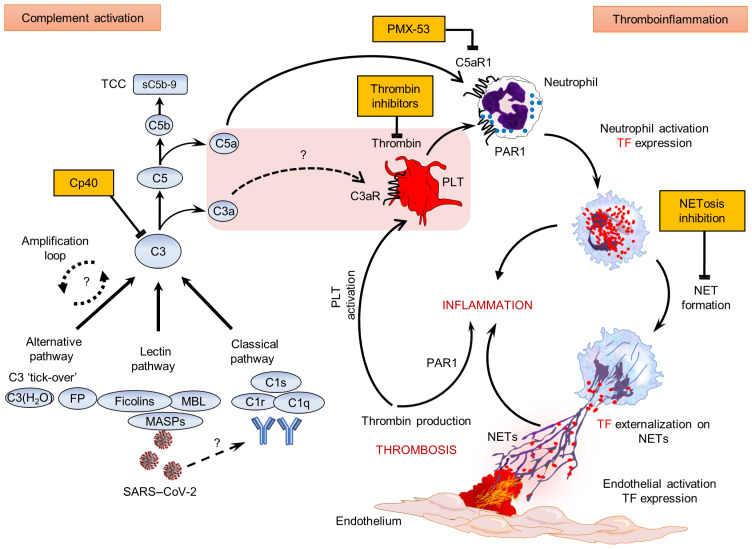
Emerging data indicate that complement and neutrophils contribute to the maladaptive immune response that fuels hyperinflammation and thrombotic microangiopathy, thereby increasing coronavirus 2019 (COVID-19) mortality. Here, we investigated how complement interacts with the platelet/neutrophil extracellular traps (NETs)/thrombin axis, using COVID-19 specimens, cell-based inhibition studies, and NET/human aortic endothelial cell (HAEC) cocultures. Increased plasma levels of NETs, tissue factor (TF) activity, and sC5b-9 were detected in patients. Neutrophils of patients yielded high TF expression and released NETs carrying active TF. Treatment of control neutrophils with COVID-19 platelet-rich plasma generated TF-bearing NETs that induced thrombotic activity of HAECs. Thrombin or NETosis inhibition or C5aR1 blockade attenuated platelet-mediated NET-driven thrombogenicity. COVID-19 serum induced complement activation in vitro, consistent with high complement activity in clinical samples. Complement C3 inhibition with compstatin Cp40 disrupted TF expression in neutrophils. In conclusion, we provide a mechanistic basis for a pivotal role of complement and NETs in COVID-19 immunothrombosis. This study supports strategies against severe acute respiratory syndrome coronavirus 2 that exploit complement or NETosis inhibition.
Keywords: COVID-19; Complement; Immunology; Neutrophils; Thrombosis.
Conflict of interest statement
Figures
Comment in
- Complement and coagulation: key triggers of COVID-19–induced multiorgan pathology
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
