

Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection
- PMID: 32759818
- PMCID: PMC7432934
- DOI: 10.3390/ijms21155585
Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection
Abstract
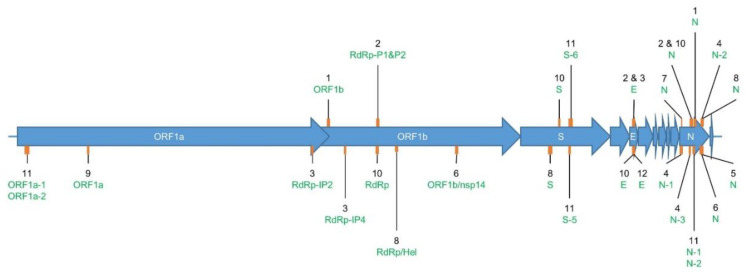
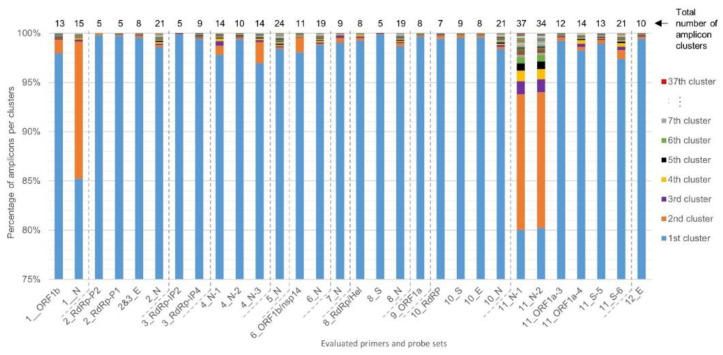
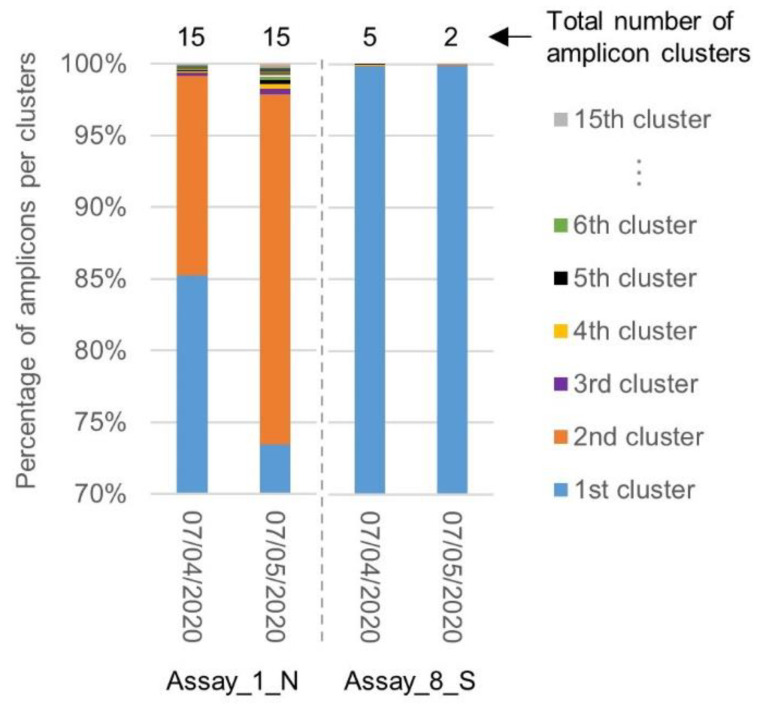
The current COronaVIrus Disease 2019 (COVID-19) pandemic started in December 2019. COVID-19 cases are confirmed by the detection of SARS-CoV-2 RNA in biological samples by RT-qPCR. However, limited numbers of SARS-CoV-2 genomes were available when the first RT-qPCR methods were developed in January 2020 for initial in silico specificity evaluation and to verify whether the targeted loci are highly conserved. Now that more whole genome data have become available, we used the bioinformatics tool SCREENED and a total of 4755 publicly available SARS-CoV-2 genomes, downloaded at two different time points, to evaluate the specificity of 12 RT-qPCR tests (consisting of a total of 30 primers and probe sets) used for SARS-CoV-2 detection and the impact of the virus' genetic evolution on four of them. The exclusivity of these methods was also assessed using the human reference genome and 2624 closely related other respiratory viral genomes. The specificity of the assays was generally good and stable over time. An exception is the first method developed by the China Center for Disease Control and prevention (CDC), which exhibits three primer mismatches present in 358 SARS-CoV-2 genomes sequenced mainly in Europe from February 2020 onwards. The best results were obtained for the assay of Chan et al. (2020) targeting the gene coding for the spiking protein (S). This demonstrates that our user-friendly strategy can be used for a first in silico specificity evaluation of future RT-qPCR tests, as well as verifying that the former methods are still capable of detecting circulating SARS-CoV-2 variants.
Keywords: COVID-19; RT-qPCR; SARS-CoV-2; WGS data; bioinformatics tool; detection; diagnosis; in silico specificity evaluation; mismatches; primers and probes.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures

References
-
- Menni C., Valdes A.M., Freidin M.B., Sudre C.H., Nguyen L.H., Drew D.A., Ganesh S., Varsavsky T., Cardoso M.J., El-Sayed Moustafa J.S., et al. Real-time tracking of self-reported symptoms to predict potential COVID-19. Nat. Med. 2020;26:1037–1040. doi: 10.1038/s41591-020-0916-2. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
