

Is the COVID-19 thrombotic catastrophe complement-connected?
- PMID: 32762081
- PMCID: PMC7436532
- DOI: 10.1111/jth.15050
Is the COVID-19 thrombotic catastrophe complement-connected?
Abstract
In December 2019, the world was introduced to a new betacoronavirus, referred to as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for its propensity to cause rapidly progressive lung damage, resulting in high death rates. As fast as the virus spread, it became evident that the novel coronavirus causes a multisystem disease (COVID-19) that may involve multiple organs and has a high risk of thrombosis associated with striking elevations in pro-inflammatory cytokines, D-dimer, and fibrinogen, but without disseminated intravascular coagulation. Postmortem studies have confirmed the high incidence of venous thromboembolism, but also notably revealed diffuse microvascular thrombi with endothelial swelling, consistent with a thrombotic microangiopathy, and inter-alveolar endothelial deposits of complement activation fragments. The clinicopathologic presentation of COVID-19 thus parallels that of other thrombotic diseases, such as atypical hemolytic uremic syndrome (aHUS), that are caused by dysregulation of the complement system. This raises the specter that many of the thrombotic complications arising from SARS-CoV-2 infections may be triggered and/or exacerbated by excess complement activation. This is of major potential clinical relevance, as currently available anti-complement therapies that are highly effective in protecting against thrombosis in aHUS, could be efficacious in COVID-19. In this review, we provide mounting evidence for complement participating in the pathophysiology underlying the thrombotic diathesis associated with pathogenic coronaviruses, including SARS-CoV-2. Based on current knowledge of complement, coagulation and the virus, we suggest lines of study to identify novel therapeutic targets and the rationale for clinical trials with currently available anti-complement agents for COVID-19.
Keywords: complement; covid-19; microvascular; thrombotic microangiopathy; tissue factor.
© 2020 International Society on Thrombosis and Haemostasis.
Figures
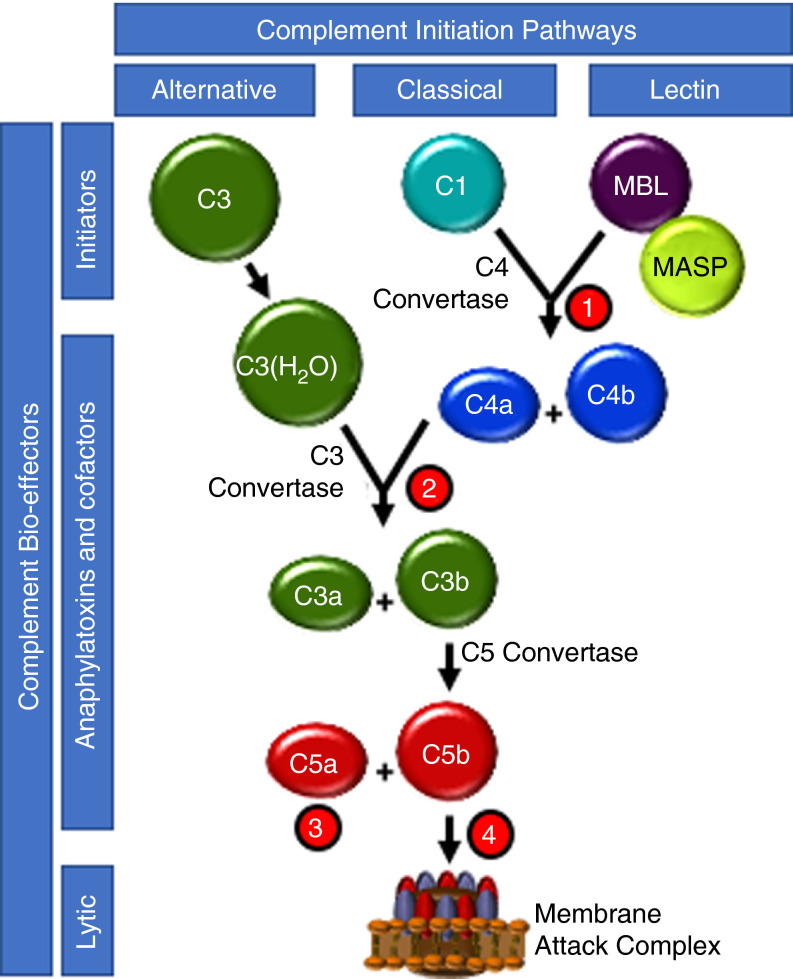
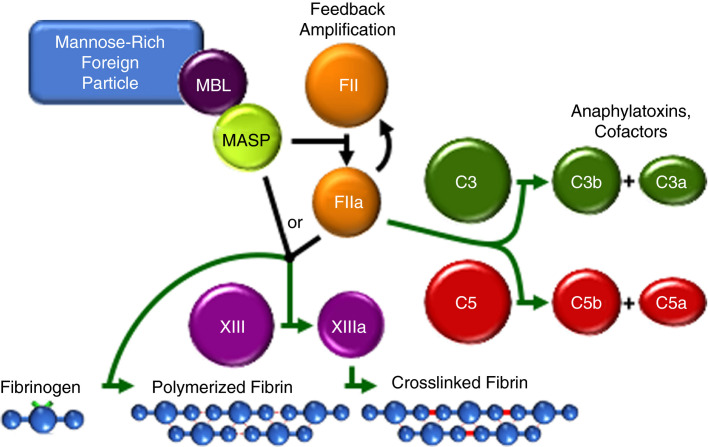
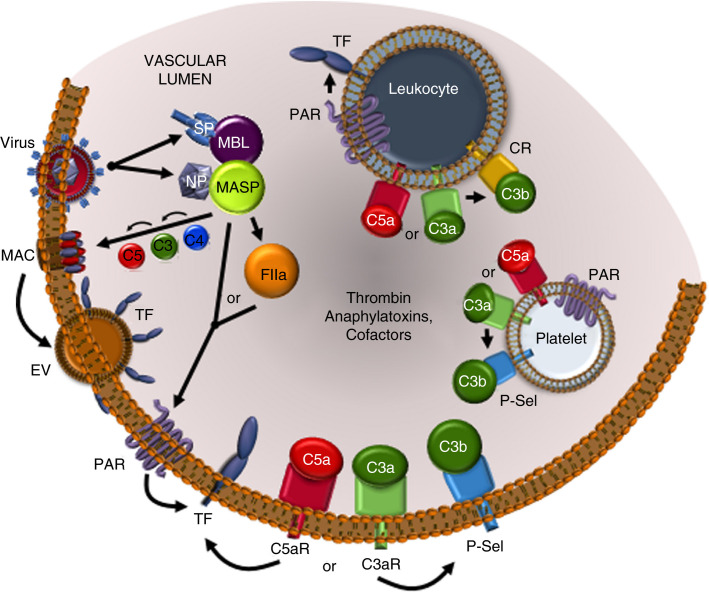
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
