

Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology
- PMID: 32764324
- PMCID: PMC7463646
- DOI: 10.3390/cells9081838
Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology
Abstract
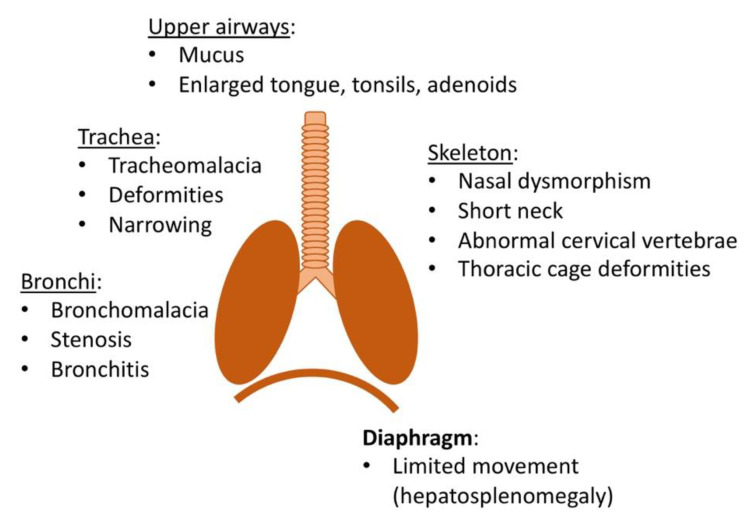
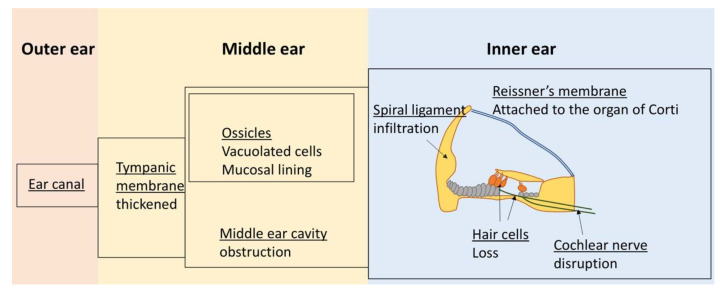
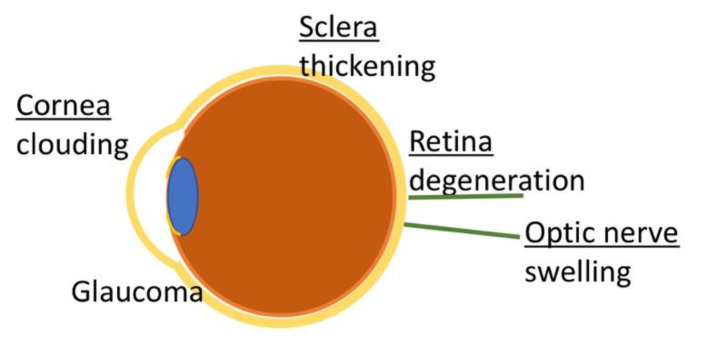
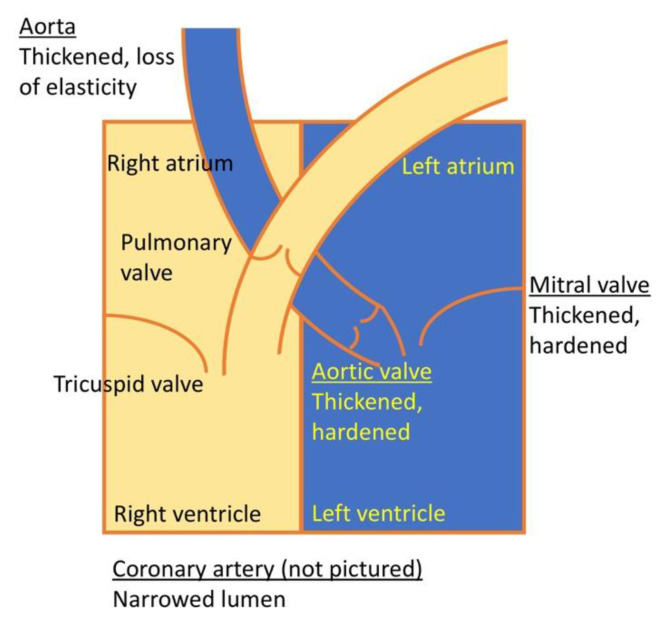
Mucopolysaccharidosis type I (MPS I) is a rare autosomal recessive inherited disease, caused by deficiency of the enzyme α-L-iduronidase, resulting in accumulation of the glycosaminoglycans (GAGs) dermatan and heparan sulfate in organs and tissues. If untreated, patients with the severe phenotype die within the first decade of life. Early diagnosis is crucial to prevent the development of fatal disease manifestations, prominently cardiac and respiratory disease, as well as cognitive impairment. However, the initial symptoms are nonspecific and impede early diagnosis. This review discusses common phenotypic manifestations in the order in which they develop. Similarities and differences in the three animal models for MPS I are highlighted. Earliest symptoms, which present during the first 6 months of life, include hernias, coarse facial features, recurrent rhinitis and/or upper airway obstructions in the absence of infection, and thoracolumbar kyphosis. During the next 6 months, loss of hearing, corneal clouding, and further musculoskeletal dysplasias develop. Finally, late manifestations including lower airway obstructions and cognitive decline emerge. Cardiac symptoms are common in MPS I and can develop in infancy. The underlying pathogenesis is in the intra- and extracellular accumulation of partially degraded GAGs and infiltration of cells with enlarged lysosomes causing tissue expansion and bone deformities. These interfere with the proper arrangement of collagen fibrils, disrupt nerve fibers, and cause devastating secondary pathophysiological cascades including inflammation, oxidative stress, and other disruptions to intracellular and extracellular homeostasis. A greater understanding of the natural history of MPS I will allow early diagnosis and timely management of the disease facilitating better treatment outcomes.
Keywords: animal models; mucopolysaccharidosis type I; α-L-iduronidase.
Conflict of interest statement
C.S.H., M.S., J.W. and R.S.M. are employees of Immusoft Corporation. T.C.L. is a paid consultant of Immusoft Corporation.
Figures
Similar articles
-
Mucopolysaccharidosis type I.Pediatr Endocrinol Rev. 2014 Sep;12 Suppl 1:102-6. Pediatr Endocrinol Rev. 2014. PMID: 25345091 Review.
-
Differences in MPS I and MPS II Disease Manifestations.Int J Mol Sci. 2021 Jul 23;22(15):7888. doi: 10.3390/ijms22157888. Int J Mol Sci. 2021. PMID: 34360653 Free PMC article. Review.
-
Replacing the enzyme alpha-L-iduronidase at birth ameliorates symptoms in the brain and periphery of dogs with mucopolysaccharidosis type I.Sci Transl Med. 2010 Dec 1;2(60):60ra89. doi: 10.1126/scitranslmed.3001380. Sci Transl Med. 2010. PMID: 21123810 Free PMC article.
-
A common mutation for mucopolysaccharidosis type I associated with a severe Hurler syndrome phenotype.Hum Mutat. 1992;1(2):103-8. doi: 10.1002/humu.1380010204. Hum Mutat. 1992. PMID: 1301196
-
Diagnosing lysosomal storage disorders: mucopolysaccharidosis type I.Curr Protoc Hum Genet. 2015 Jan 20;84:17.17.1-17.17.8. doi: 10.1002/0471142905.hg1717s84. Curr Protoc Hum Genet. 2015. PMID: 25599668
Cited by
-
Efficacy of a Combination Therapy with Laronidase and Genistein in Treating Mucopolysaccharidosis Type I in a Mouse Model.Int J Mol Sci. 2024 Feb 17;25(4):2371. doi: 10.3390/ijms25042371. Int J Mol Sci. 2024. PMID: 38397051 Free PMC article.
-
Enzyme replacement with transferrin receptor-targeted α-L-iduronidase rescues brain pathology in mucopolysaccharidosis I mice.Mol Ther Methods Clin Dev. 2023 May 12;29:439-449. doi: 10.1016/j.omtm.2023.05.010. eCollection 2023 Jun 8. Mol Ther Methods Clin Dev. 2023. PMID: 37251981 Free PMC article.
-
Efficacy and safety of a biosimilar laronidase versus the reference laronidase in patients with mucopolysaccharidosis type I.Sci Rep. 2025 Aug 19;15(1):30427. doi: 10.1038/s41598-025-16351-4. Sci Rep. 2025. PMID: 40830667 Free PMC article. Clinical Trial.
-
In vivo adenine base editing corrects newborn murine model of Hurler syndrome.Mol Biomed. 2023 Feb 23;4(1):6. doi: 10.1186/s43556-023-00120-8. Mol Biomed. 2023. PMID: 36813914 Free PMC article.
-
Liver-directed gene therapy corrects neurologic disease in a murine model of mucopolysaccharidosis type I-Hurler.Mol Ther Methods Clin Dev. 2022 Apr 19;25:370-381. doi: 10.1016/j.omtm.2022.04.010. eCollection 2022 Jun 9. Mol Ther Methods Clin Dev. 2022. PMID: 35573046 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
