

Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites
- PMID: 32769985
- PMCID: PMC7414224
- DOI: 10.1038/s41467-020-17858-2
Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites
Abstract
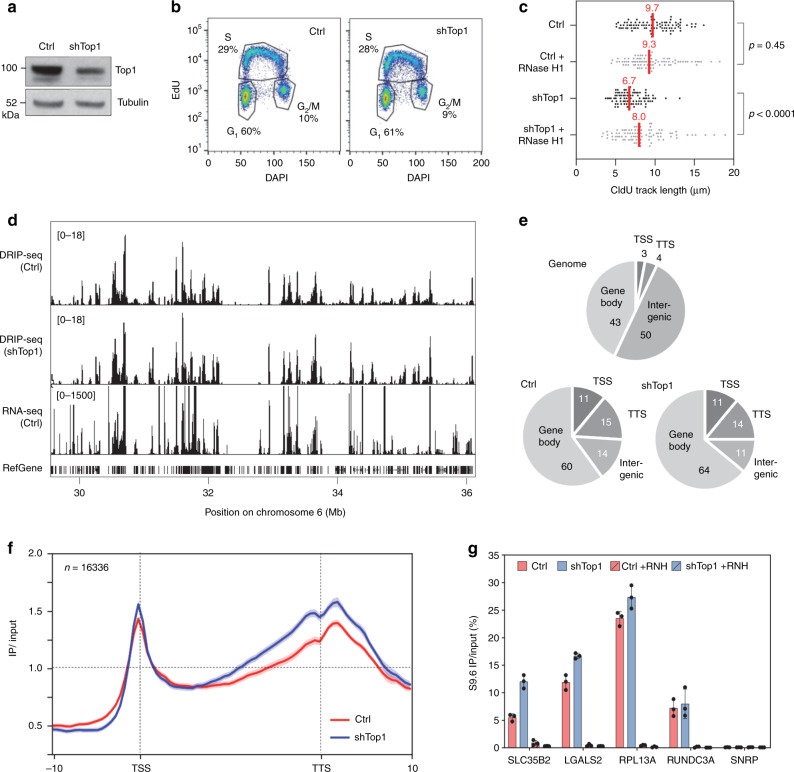
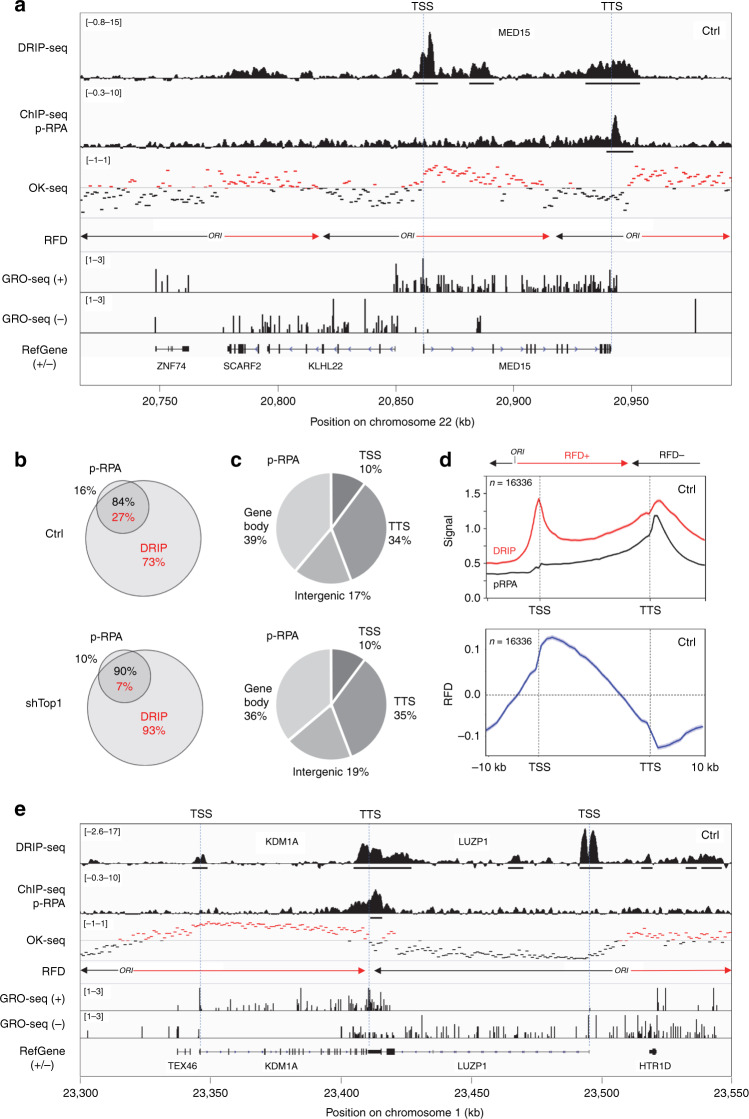
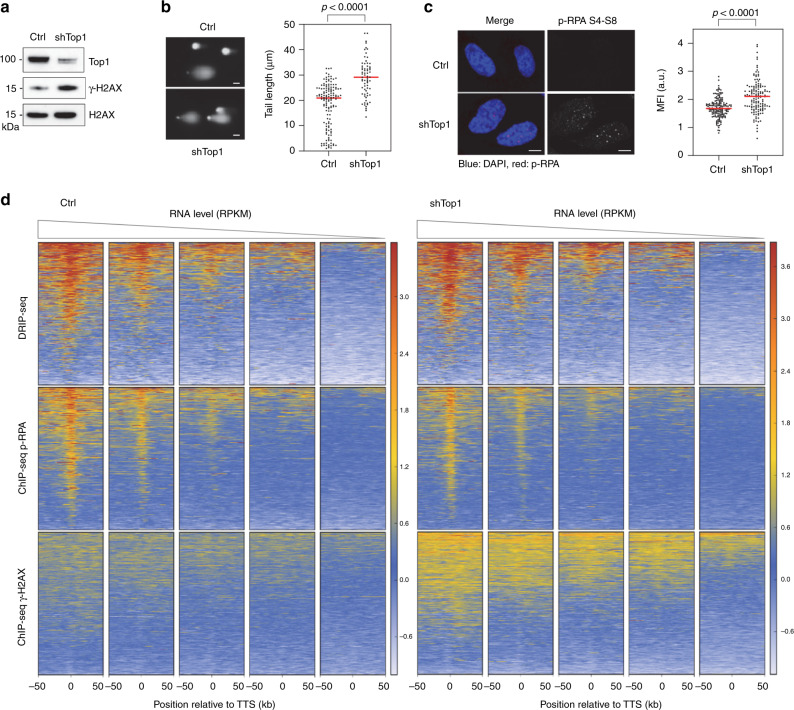
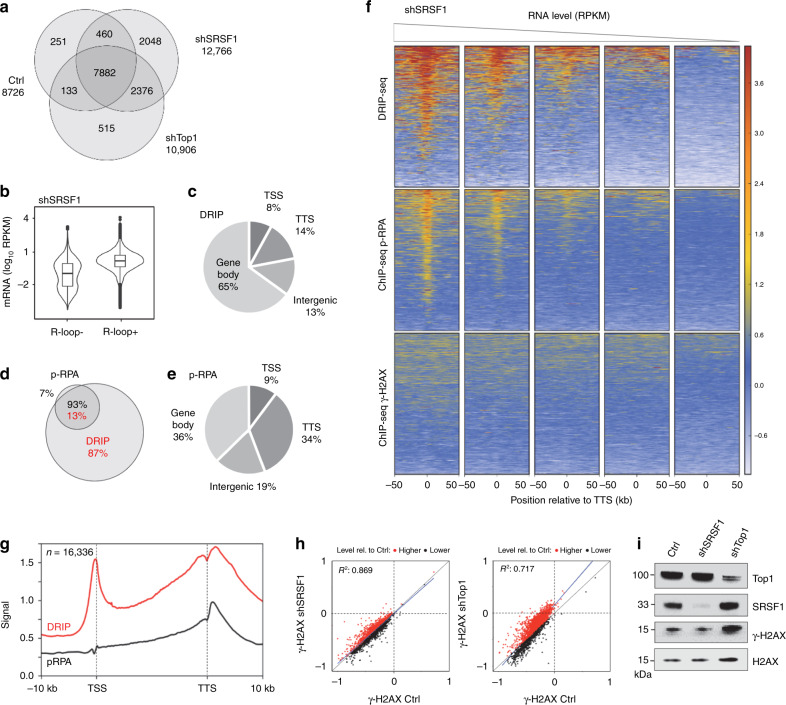
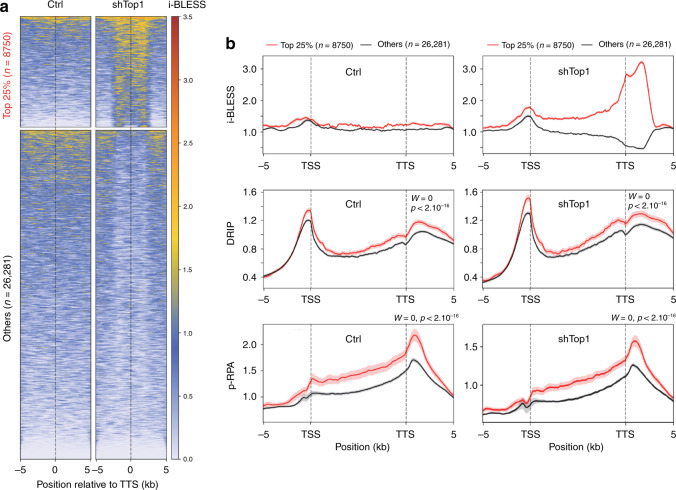
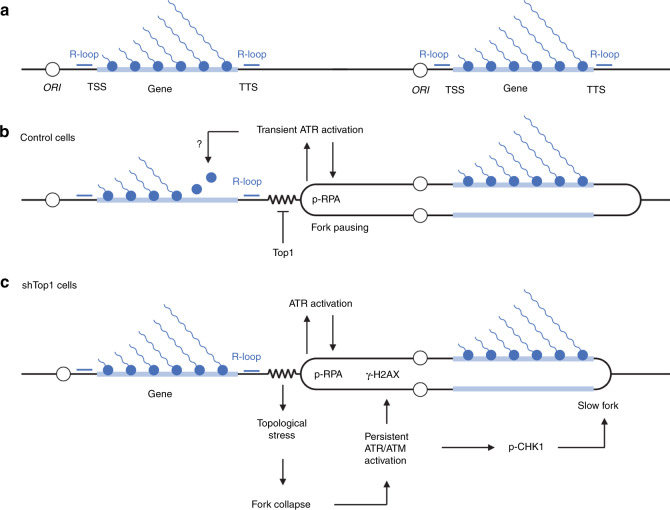
R-loops have both positive and negative impacts on chromosome functions. To identify toxic R-loops in the human genome, here, we map RNA:DNA hybrids, replication stress markers and DNA double-strand breaks (DSBs) in cells depleted for Topoisomerase I (Top1), an enzyme that relaxes DNA supercoiling and prevents R-loop formation. RNA:DNA hybrids are found at both promoters (TSS) and terminators (TTS) of highly expressed genes. In contrast, the phosphorylation of RPA by ATR is only detected at TTS, which are preferentially replicated in a head-on orientation relative to the direction of transcription. In Top1-depleted cells, DSBs also accumulate at TTS, leading to persistent checkpoint activation, spreading of γ-H2AX on chromatin and global replication fork slowdown. These data indicate that fork pausing at the TTS of highly expressed genes containing R-loops prevents head-on conflicts between replication and transcription and maintains genome integrity in a Top1-dependent manner.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015;10:425–448. - PubMed
-
- Fragkos M, Ganier O, Coulombe P, Mechali M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015;16:360–374. - PubMed
-
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. - PubMed
-
- Pasero P, Vindigni A. Nucleases acting at stalled forks: how to reboot the replication program with a few shortcuts. Annu. Rev. Genet. 2017;51:477–499. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
