

Epigenomic and Transcriptomic Dynamics During Human Heart Organogenesis
- PMID: 32772801
- PMCID: PMC7554226
- DOI: 10.1161/CIRCRESAHA.120.316704
Epigenomic and Transcriptomic Dynamics During Human Heart Organogenesis
Abstract
Rationale: There is growing evidence that common variants and rare sequence alterations in regulatory sequences can result in birth defects or predisposition to disease. Congenital heart defects are the most common birth defect and have a clear genetic component, yet only a third of cases can be attributed to structural variation in the genome or a mutation in a gene. The remaining unknown cases could be caused by alterations in regulatory sequences.
Objective: Identify regulatory sequences and gene expression networks that are active during organogenesis of the human heart. Determine whether these sites and networks are enriched for disease-relevant genes and associated genetic variation.
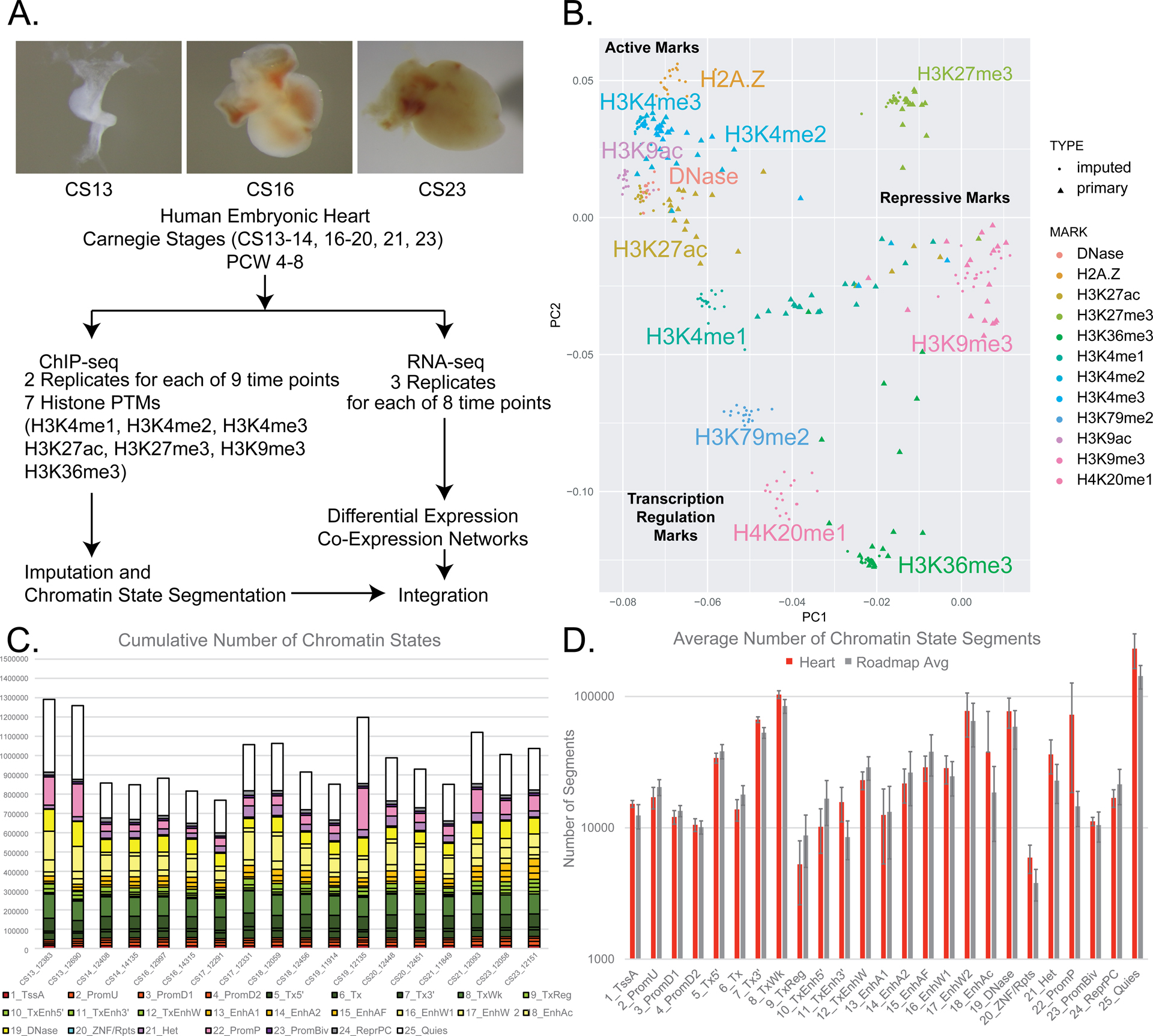
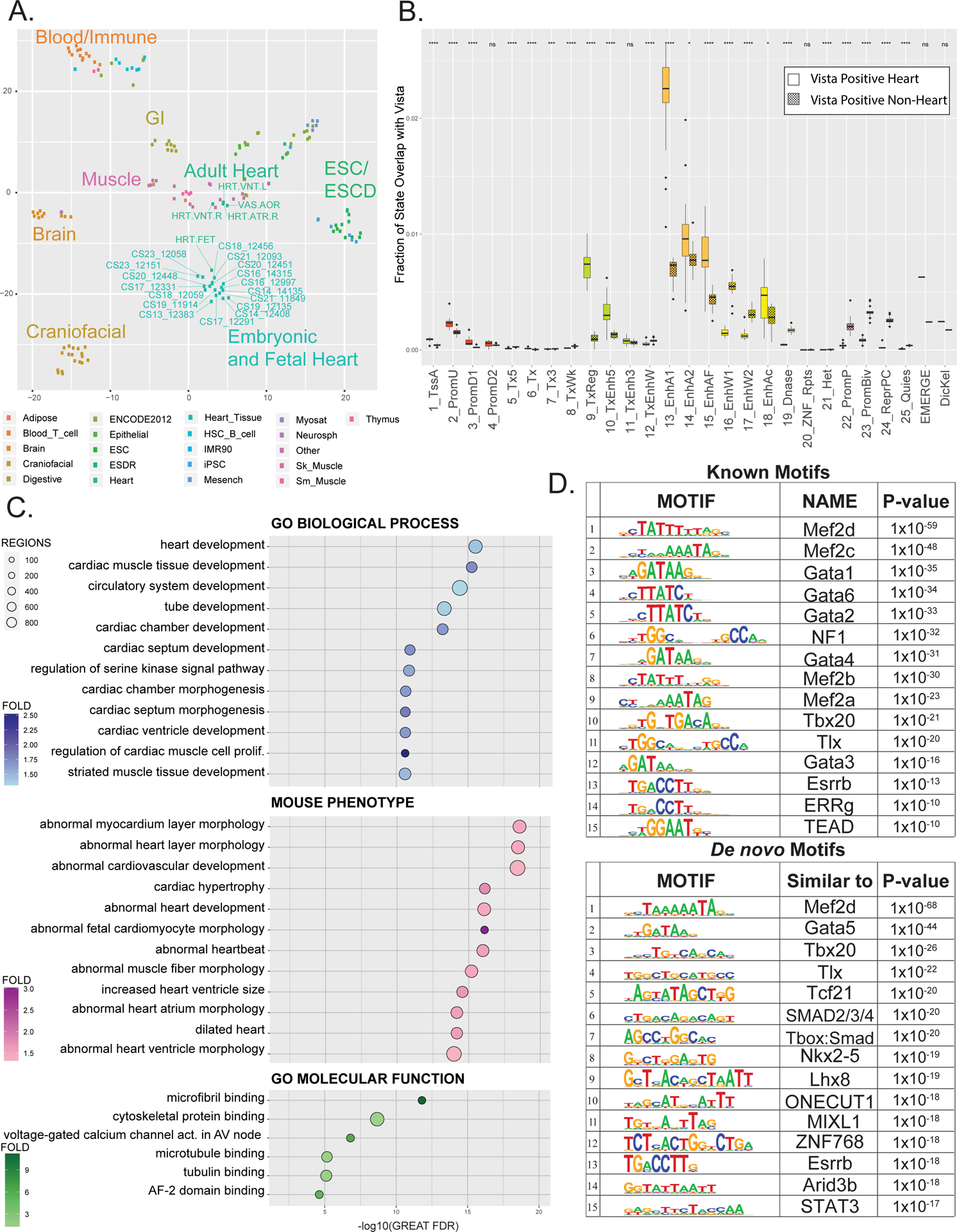
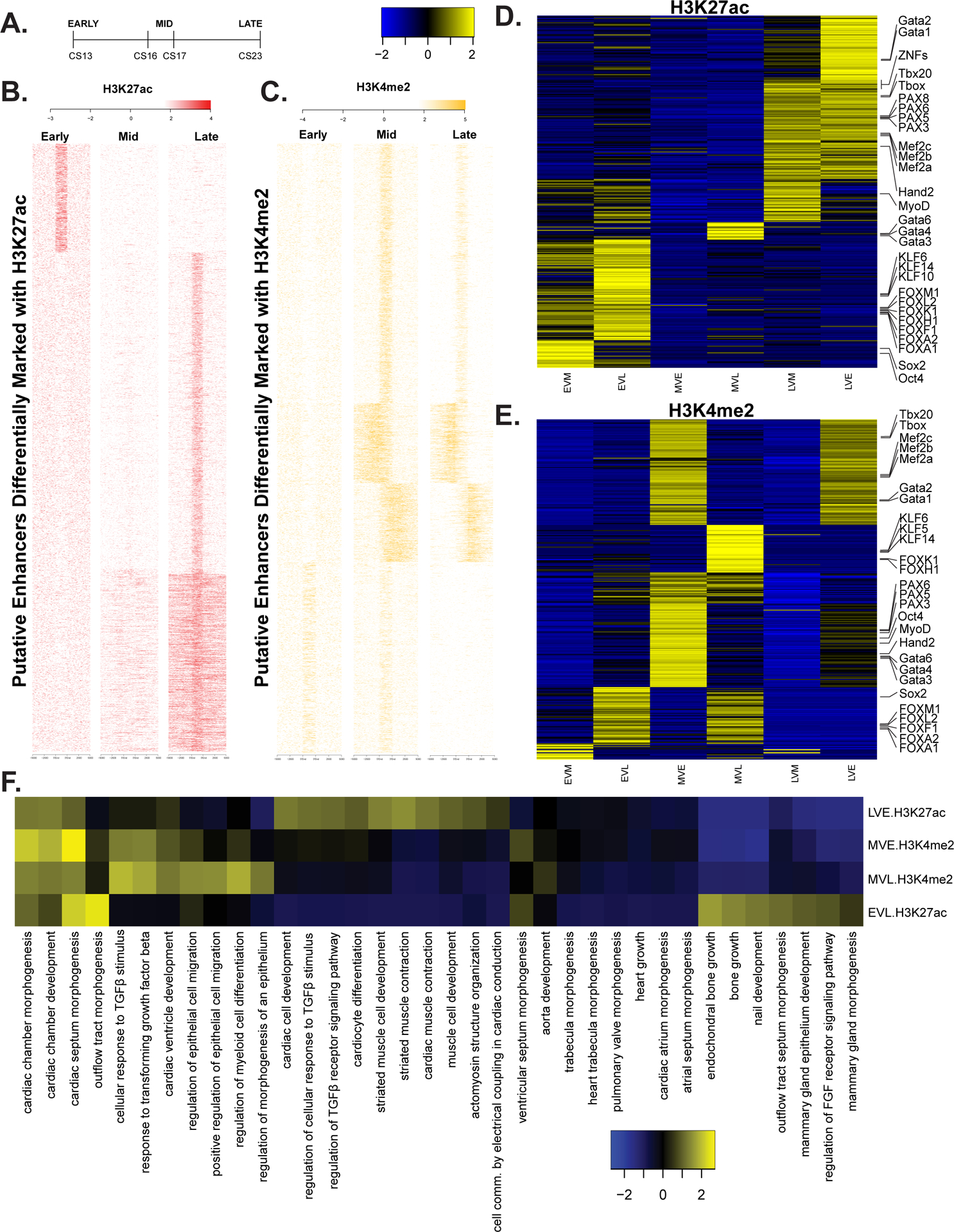
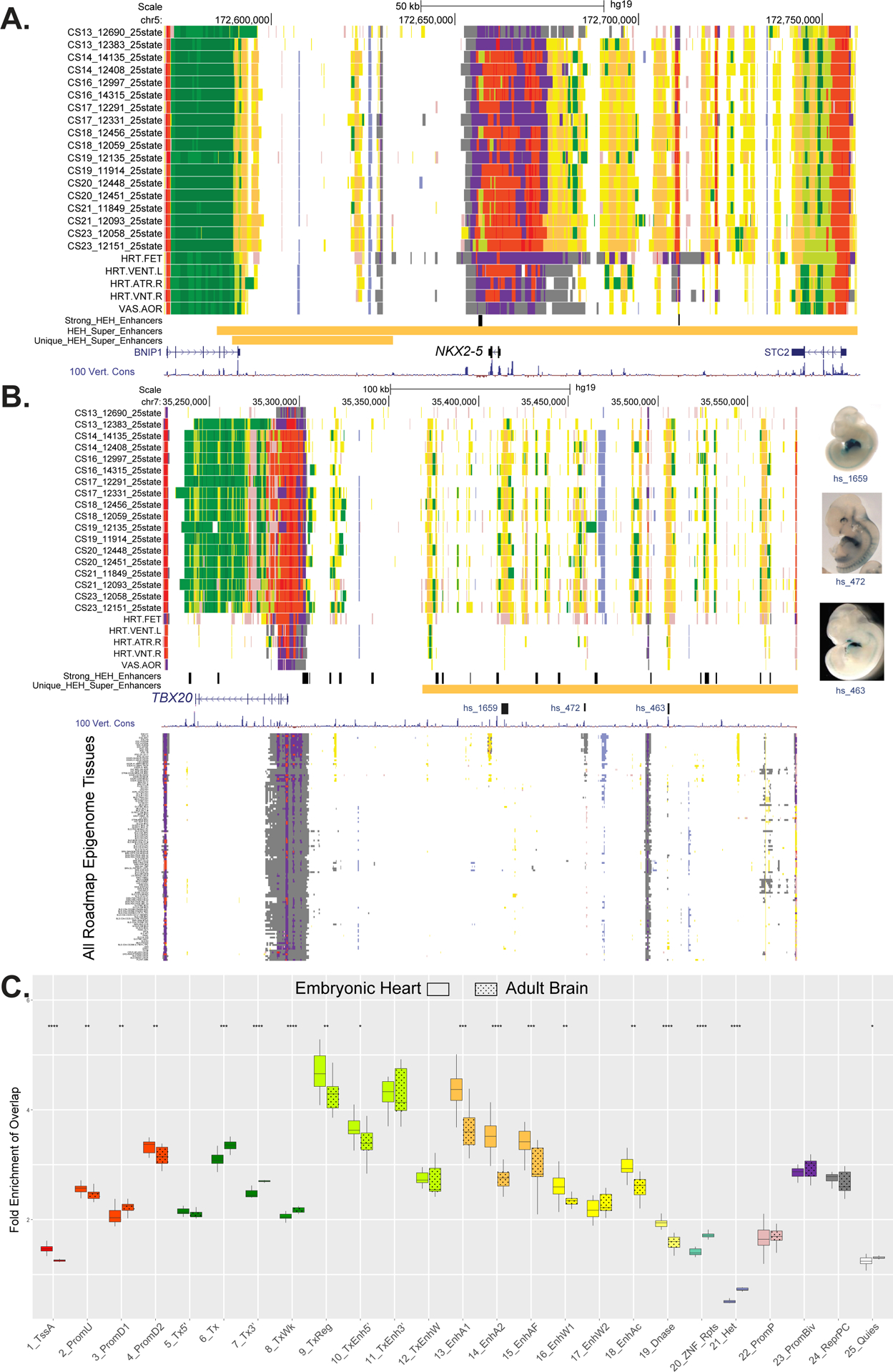
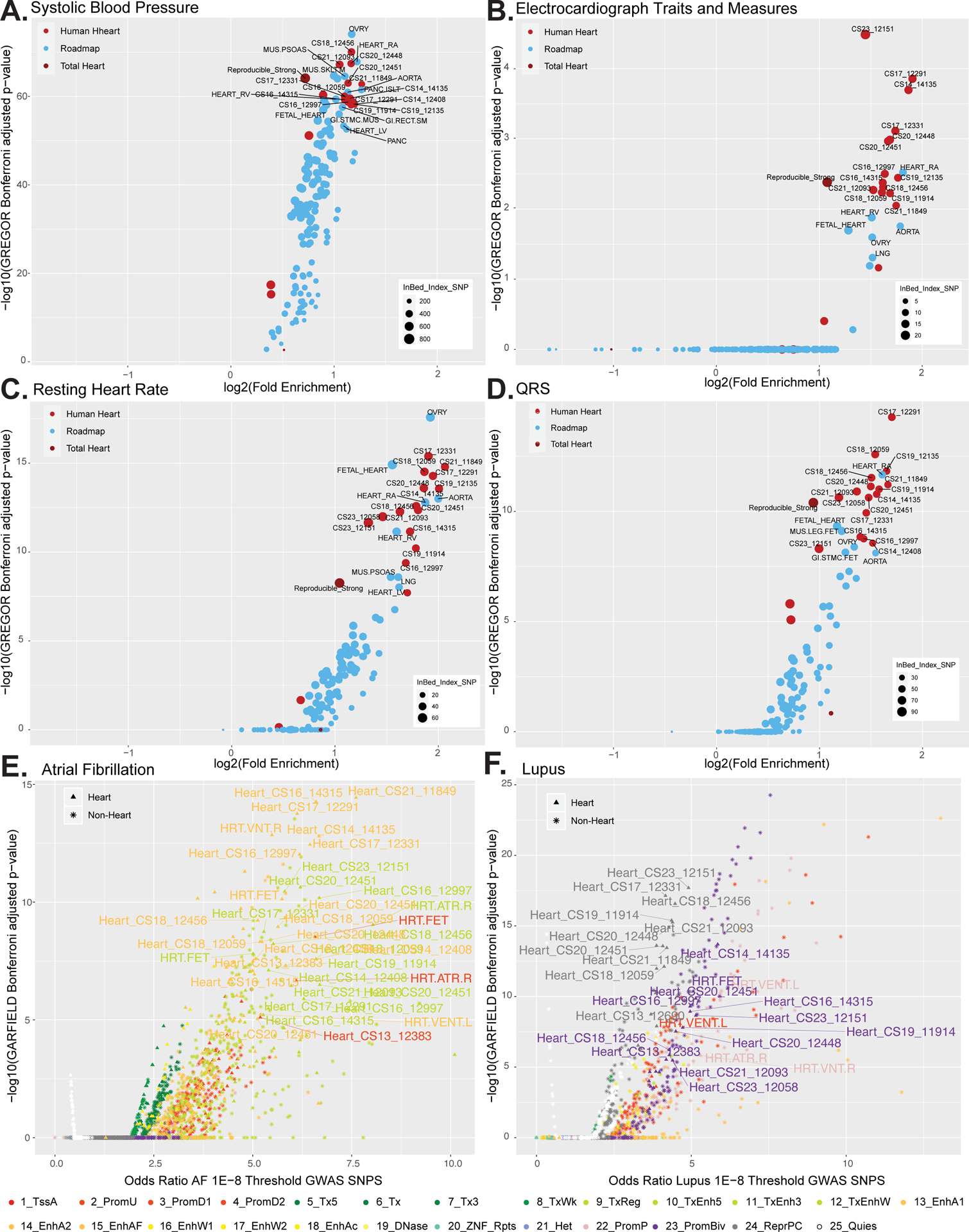
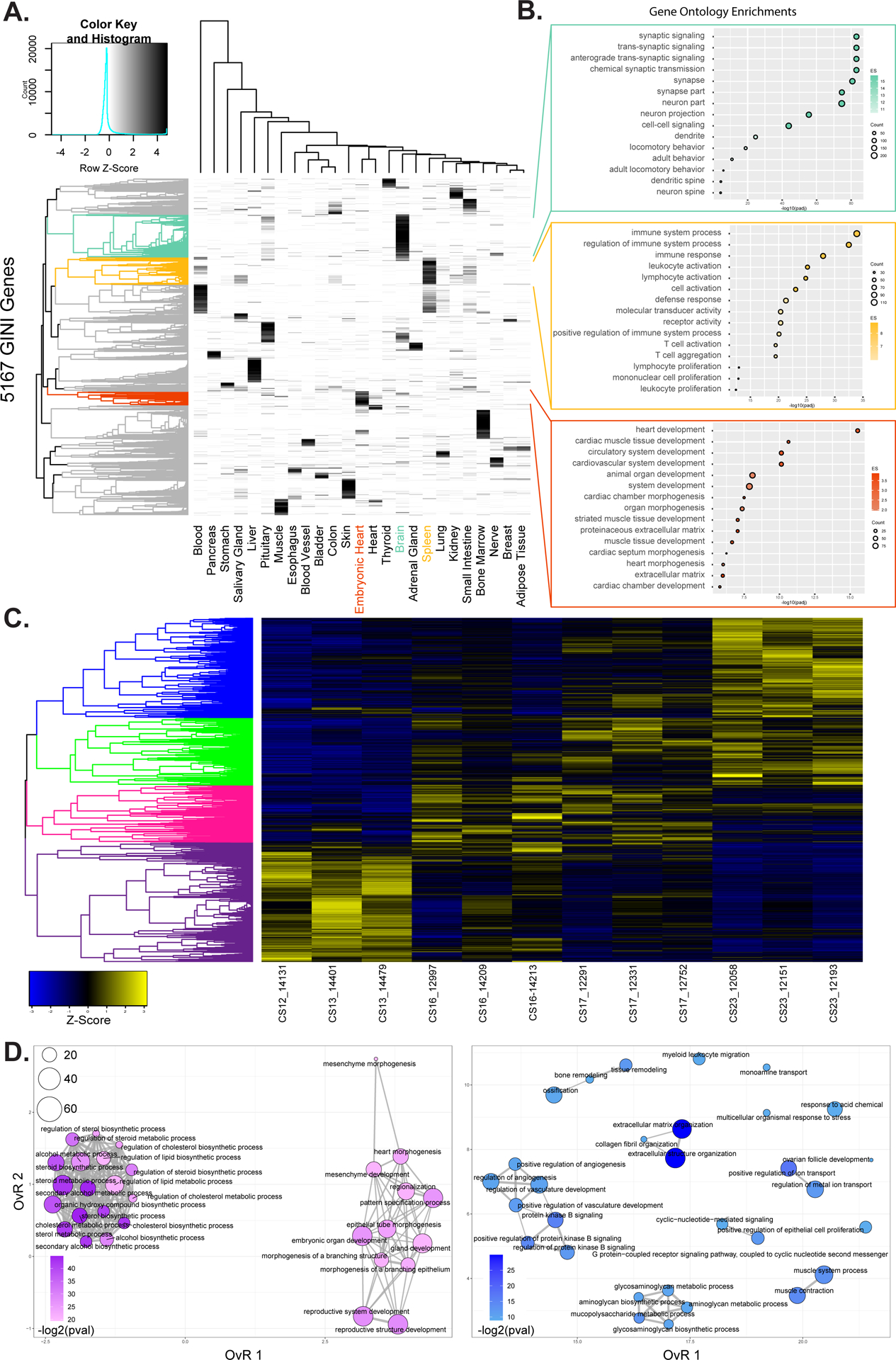
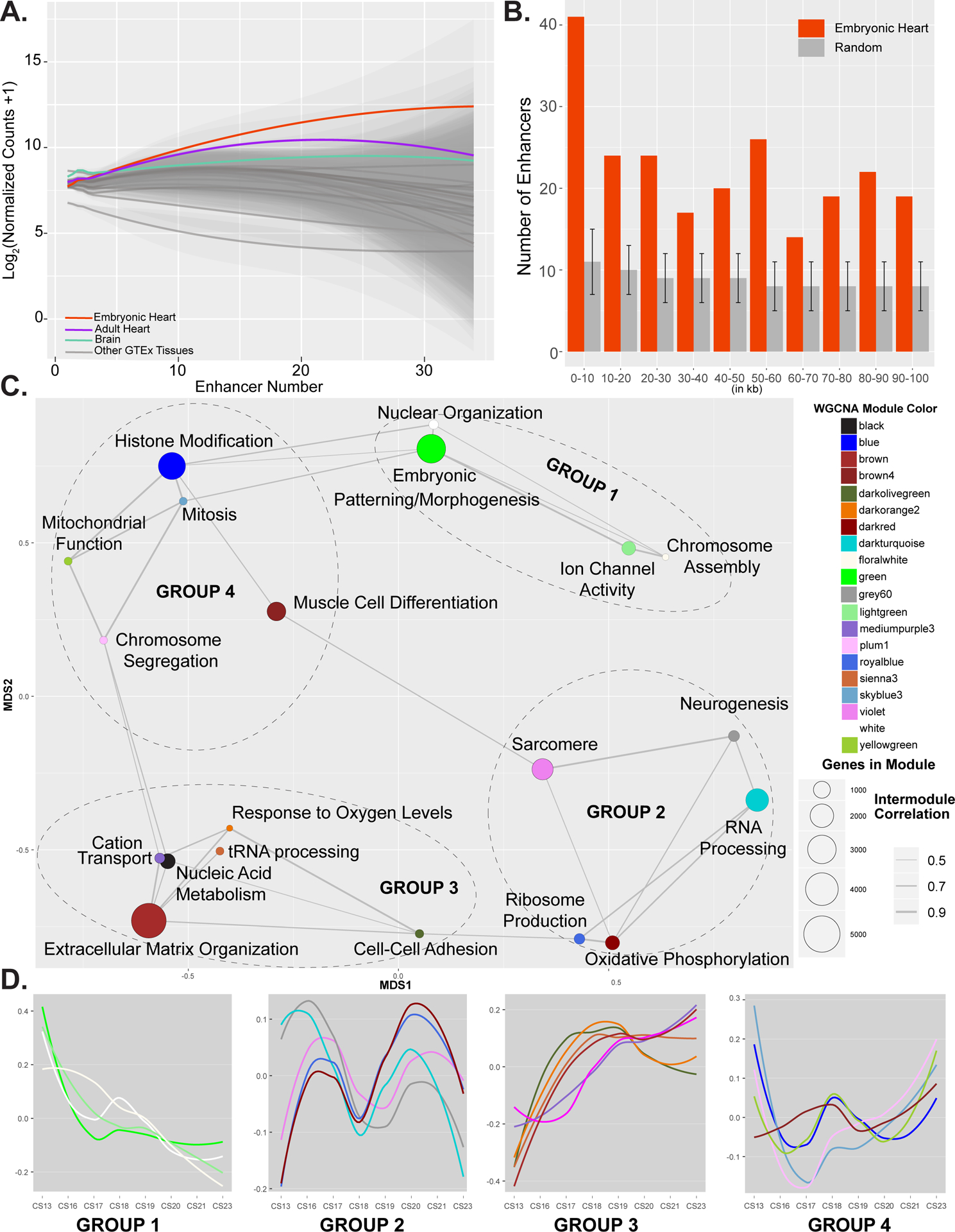
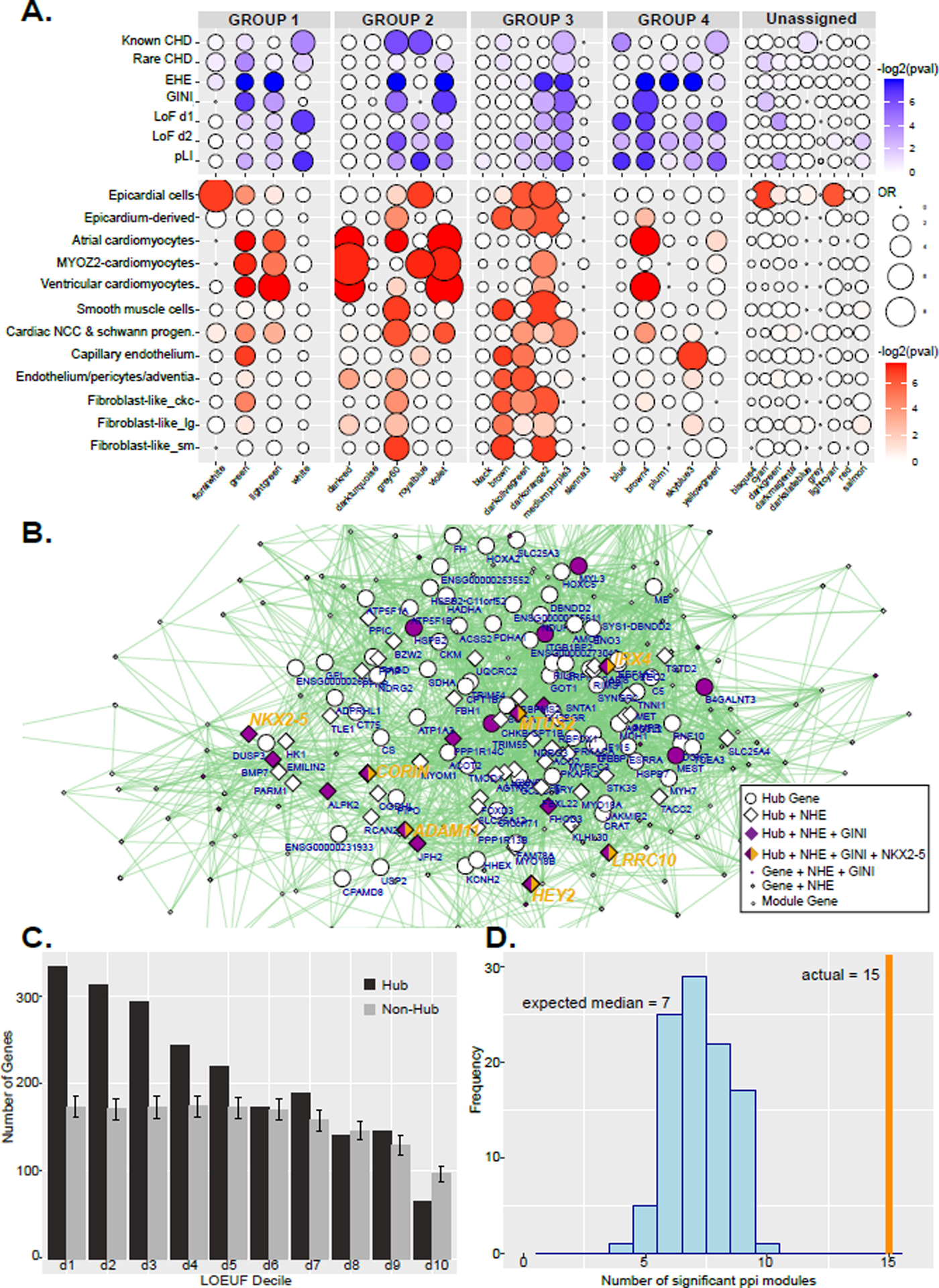
Methods and results: We characterized ChromHMM (chromatin state) and gene expression dynamics during human heart organogenesis. We profiled 7 histone modifications in embryonic hearts from each of 9 distinct Carnegie stages (13-14, 16-21, and 23), annotated chromatin states, and compared these maps to over 100 human tissues and cell types. We also generated RNA-sequencing data, performed differential expression, and constructed weighted gene coexpression networks. We identified 177 412 heart enhancers; 12 395 had not been previously annotated as strong enhancers. We identified 92% of all functionally validated heart-positive enhancers (n=281; 7.5× enrichment; P<2.2×10-16). Integration of these data demonstrated novel heart enhancers are enriched near genes expressed more strongly in cardiac tissue and are enriched for variants associated with ECG measures and atrial fibrillation. Our gene expression network analysis identified gene modules strongly enriched for heart-related functions, regulatory control by heart-specific enhancers, and putative disease genes.
Conclusions: Well-connected hub genes with heart-specific expression targeted by embryonic heart-specific enhancers are likely disease candidates. Our functional annotations will allow for better interpretation of whole genome sequencing data in the large number of patients affected by congenital heart defects.
Keywords: developmental biology; disease; epigenomics; genetics; genomics.
Figures
Comment in
-
Super Enhancers: Enhancing Human Cardiogenesis.Circ Res. 2020 Oct 9;127(9):1156-1158. doi: 10.1161/CIRCRESAHA.120.318000. Epub 2020 Oct 8. Circ Res. 2020. PMID: 33031028 Free PMC article. No abstract available.
References
-
- Van Der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol 2011;58:2241–2247. - PubMed
-
- Liu Y, Chen S, Zühlke L, Black GC, Choy M-K, Li N, Keavney BD. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol [Internet]. 2019. [cited 2019 Nov 15];48:455–463. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30783674 - PMC - PubMed
-
- Centers for Disease Control and Prevention (CDC). Racial differences by gestational age in neonatal deaths attributable to congenital heart defects --- United States, 2003–2006. MMWR Morb Mortal Wkly Rep [Internet]. 2010. [cited 2019 Nov 15];59:1208–11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20864921 - PubMed
-
- Cowan JR, Ware SM. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol 2015;42:373–393. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
