

Precision medicine and therapies of the future
- PMID: 32776321
- PMCID: PMC8432144
- DOI: 10.1111/epi.16539
Precision medicine and therapies of the future
Abstract
Precision medicine in the epilepsies has gathered much attention, especially with gene discovery pushing forward new understanding of disease biology. Several targeted treatments are emerging, some with considerable sophistication and individual-level tailoring. There have been rare achievements in improving short-term outcomes in a few very select patients with epilepsy. The prospects for further targeted, repurposed, or novel treatments seem promising. Along with much-needed success, difficulties are also arising. Precision treatments do not always work, and sometimes are inaccessible or do not yet exist. Failures of precision medicine may not find their way to broader scrutiny. Precision medicine is not a new concept: It has been boosted by genetics and is often focused on genetically determined epilepsies, typically considered to be driven in an individual by a single genetic variant. Often the mechanisms generating the full clinical phenotype from such a perceived single cause are incompletely understood. The impact of additional genetic variation and other factors that might influence the clinical presentation represent complexities that are not usually considered. Precision success and precision failure are usually equally incompletely explained. There is a need for more comprehensive evaluation and a more rigorous framework, bringing together information that is both necessary and sufficient to explain clinical presentation and clinical responses to precision treatment in a precision approach that considers the full picture not only of the effects of a single variant, but also of its genomic and other measurable environment, within the context of the whole person. As we may be on the brink of a treatment revolution, progress must be considered and reasoned: One possible framework is proposed for the evaluation of precision treatments.
Keywords: anti-seizure drugs; failure; genetics; personalized; pharmacogenetics; surgery.
© 2020 The Authors. Epilepsia published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
The author has research collaborations with UCB Pharma and Congenica Ltd. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Figures
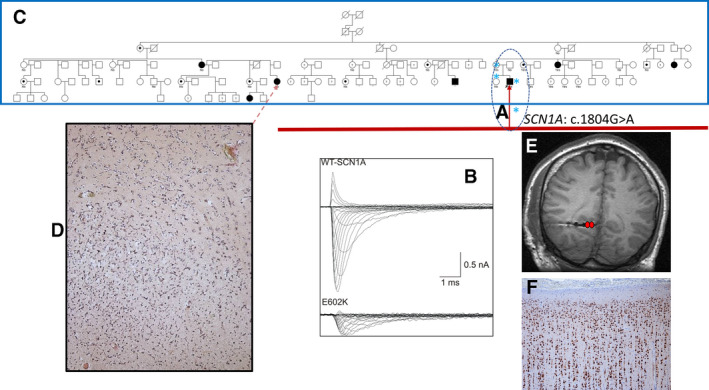
 in A, dotted oval) c.1804G>A, p.Glu602Lys was identified in the proband, his mother, and sister, and not in the asymptomatic father. Current evaluation shows that the variant absent from gnomAD, has a CADD score of 13, and a REVEL (
in A, dotted oval) c.1804G>A, p.Glu602Lys was identified in the proband, his mother, and sister, and not in the asymptomatic father. Current evaluation shows that the variant absent from gnomAD, has a CADD score of 13, and a REVEL (

References
-
- Toward precision medicine: Building a knowledge network for biomedical research and a new taxonomy of disease/Committee on A Framework for Developing a New Taxonomy of Disease, Board on Life Sciences, Division on Earth and Life Sciences, National Research Council of the National Academies. Washington, DC: National Academies Press; 2011. - PubMed
-
- Møller RS, Hammer TB, Rubboli G, Lemke JR, Johannesen KM. From next‐generation sequencing to targeted treatment of non‐acquired epilepsies. Expert Rev Mol Diagn. 2019;19(3):217–28. - PubMed
-
- Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol. 2020;19(1):93–100. - PubMed
-
- Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN2A‐related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia. 2019;60(suppl 3):S59–67. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
