

Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers
- PMID: 32778843
- PMCID: PMC8299533
- DOI: 10.1038/s41589-020-0613-y
Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers
Abstract
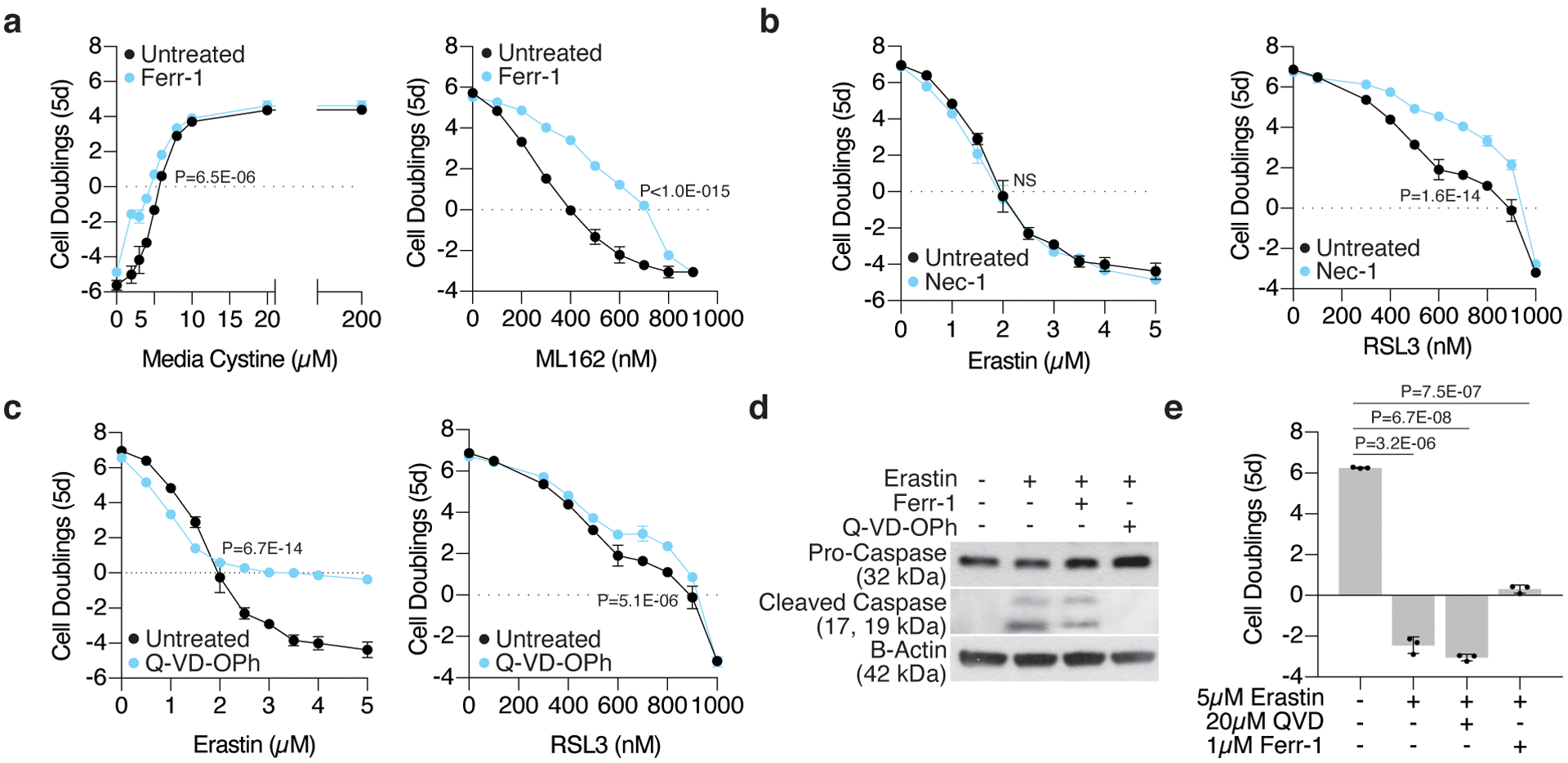
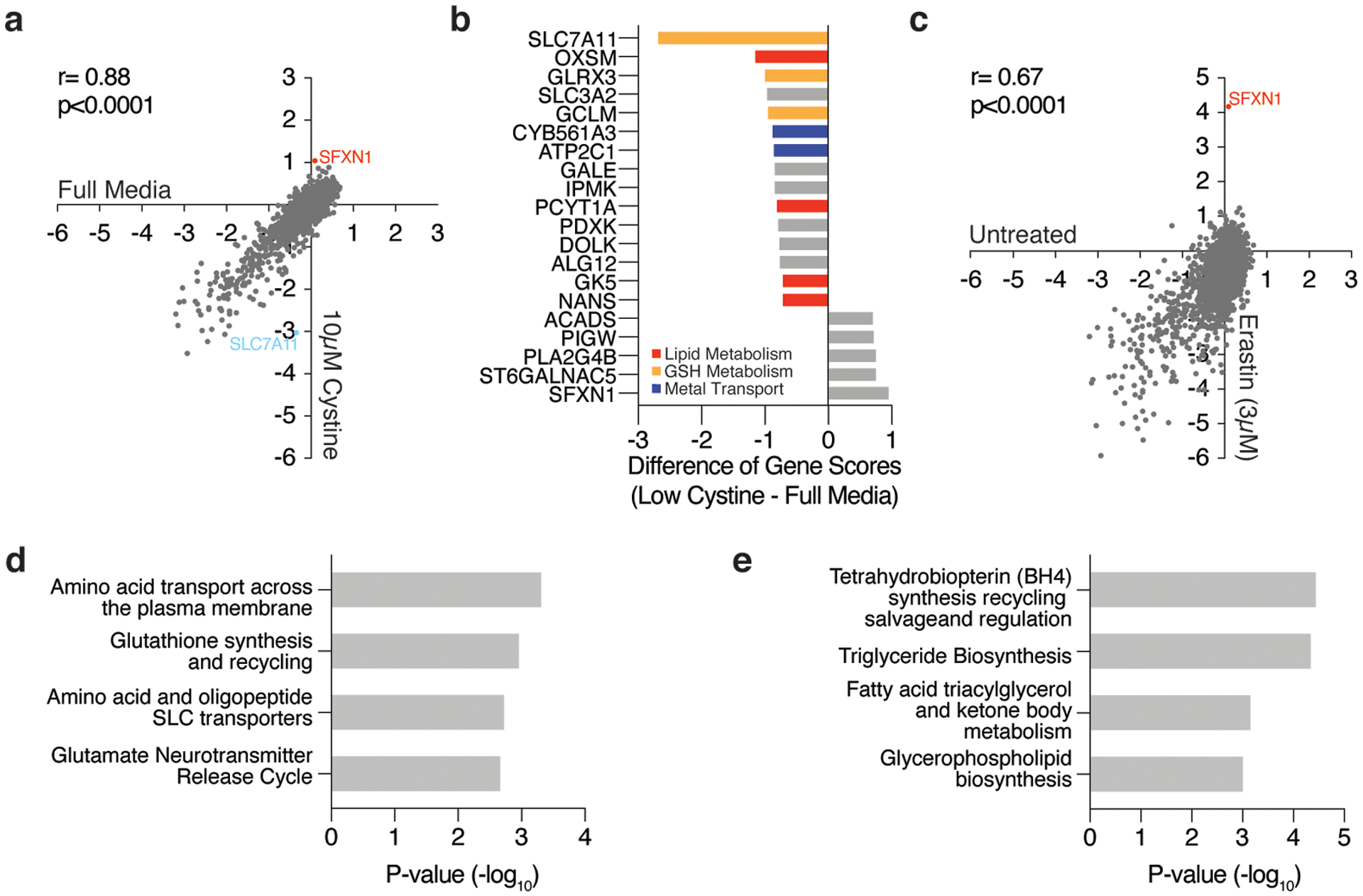
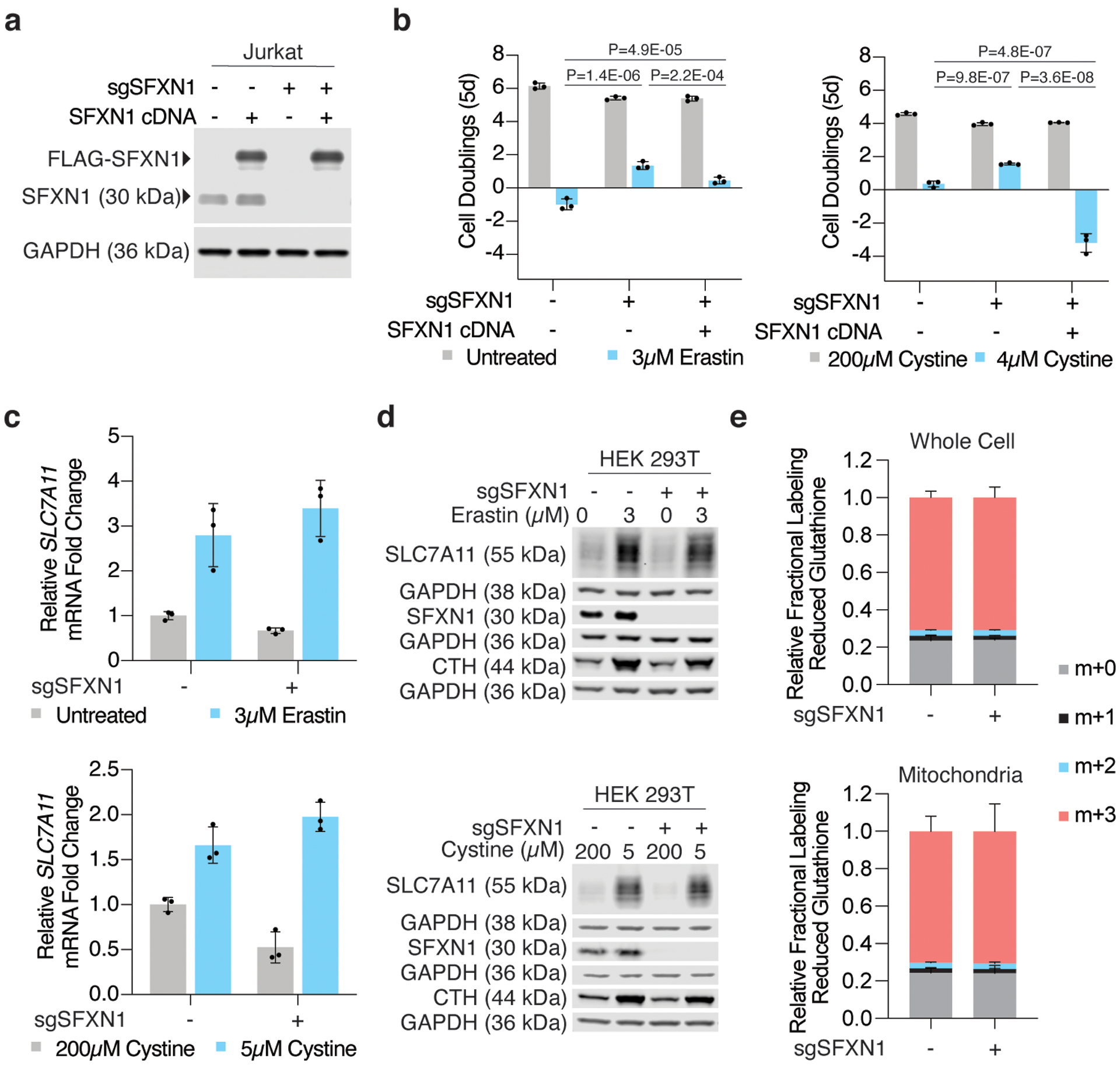
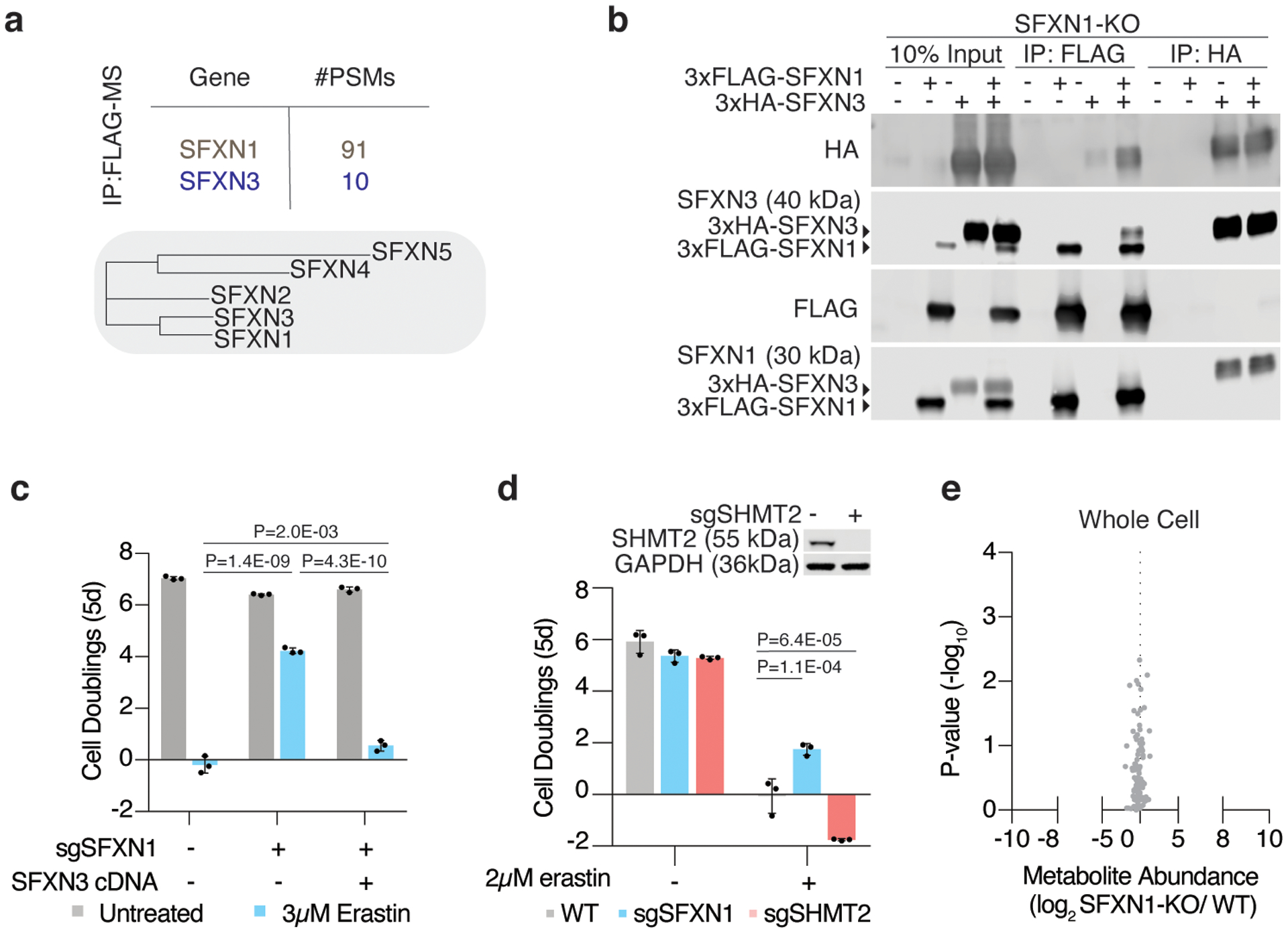
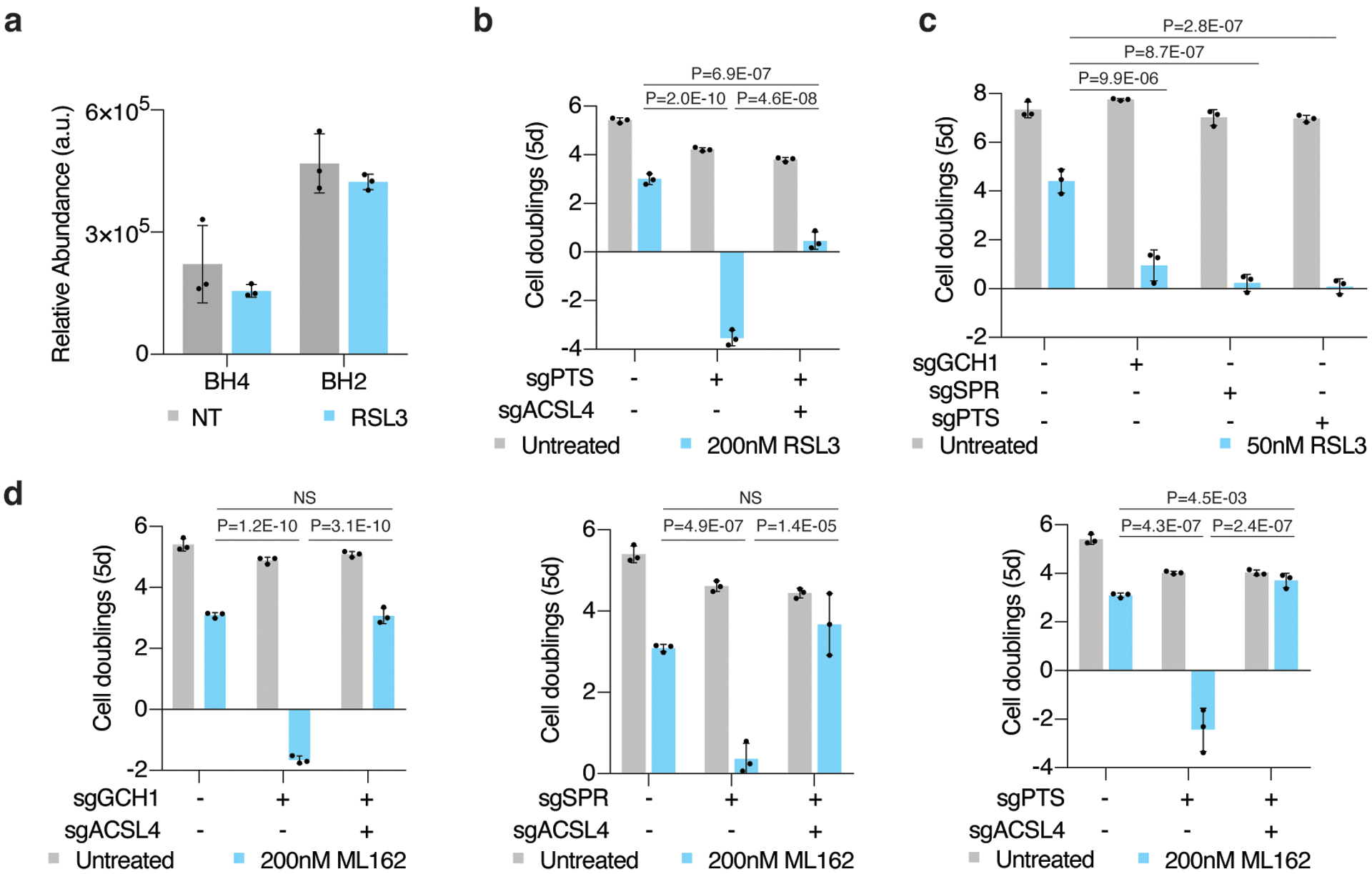
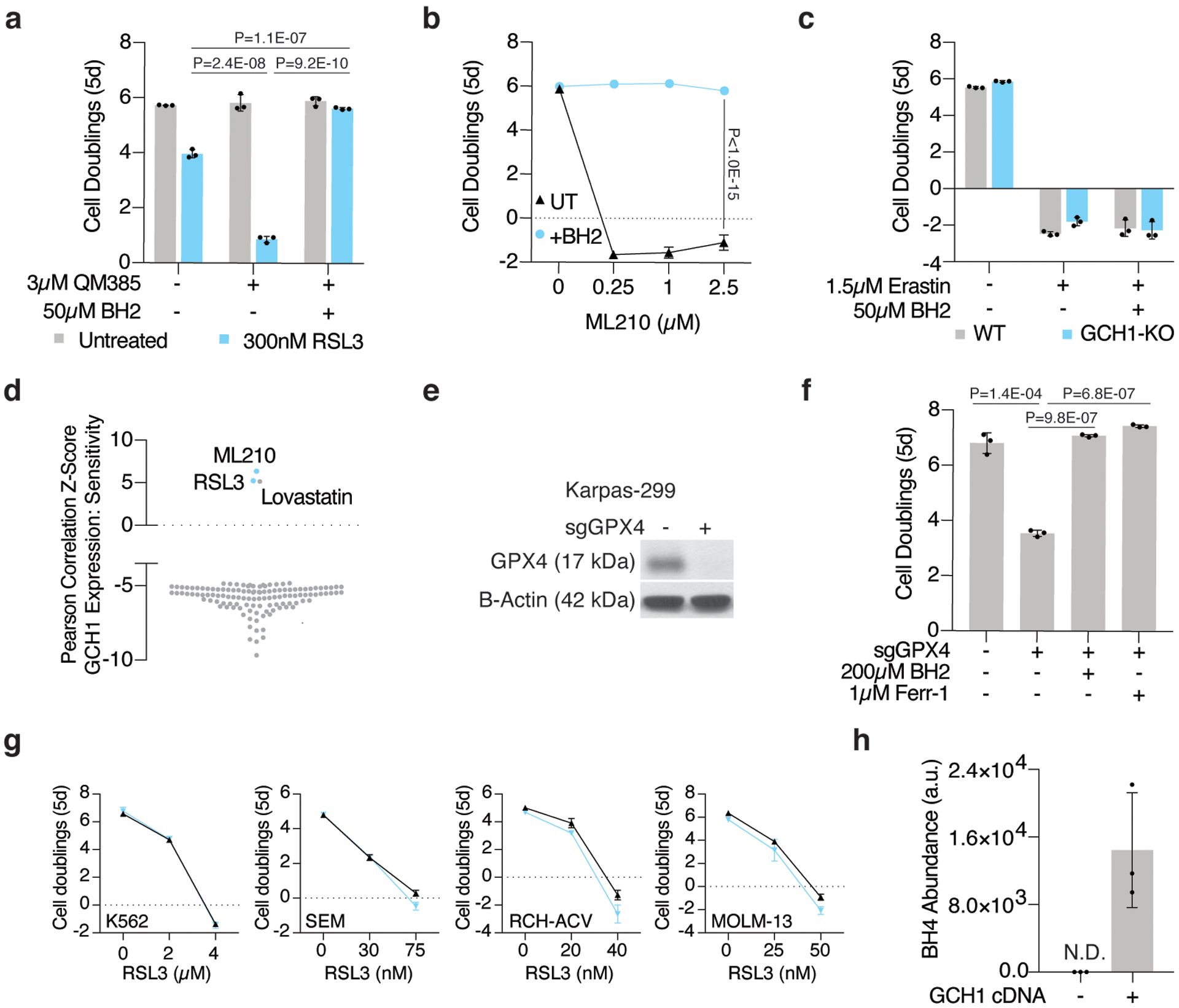
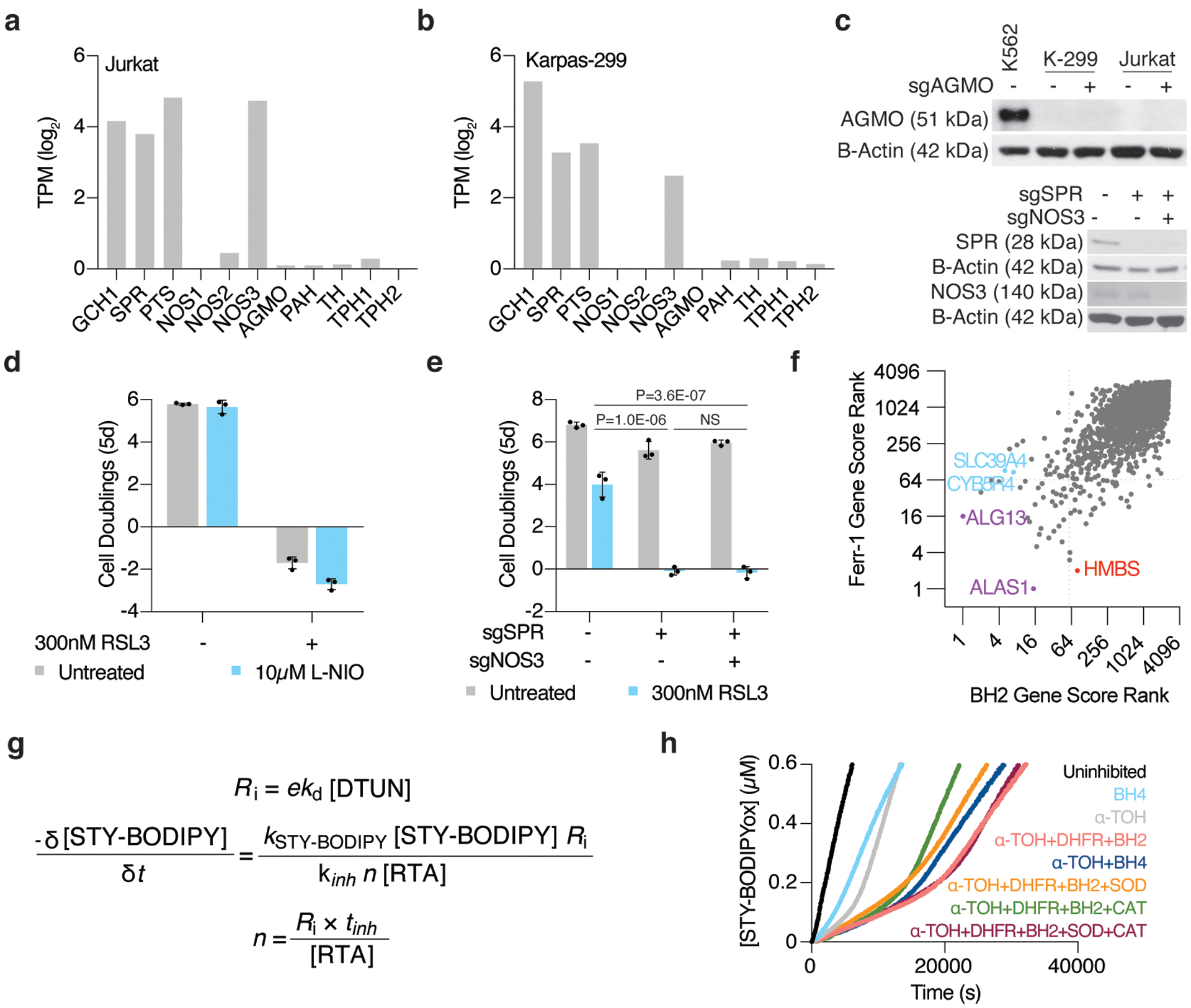
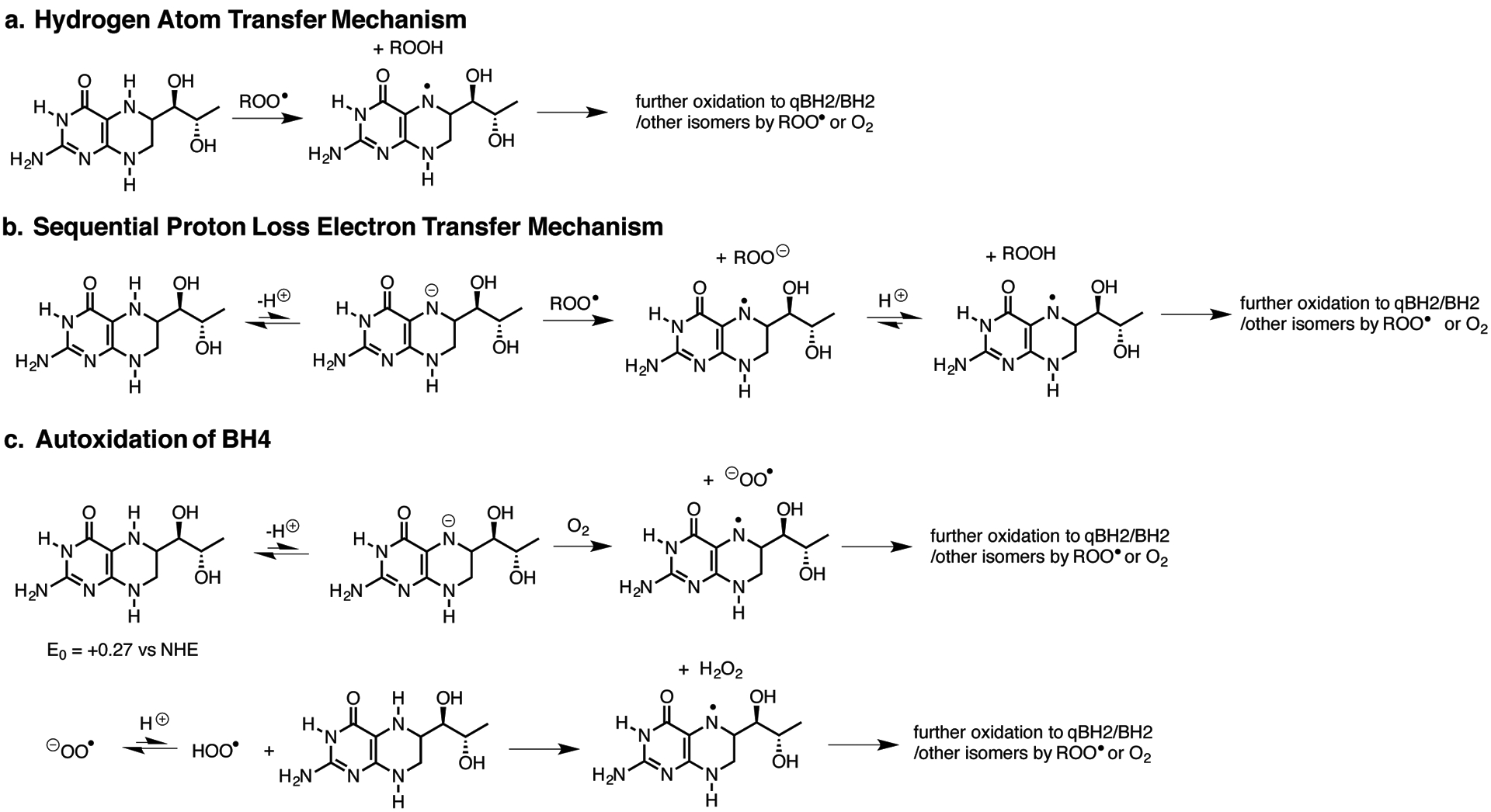
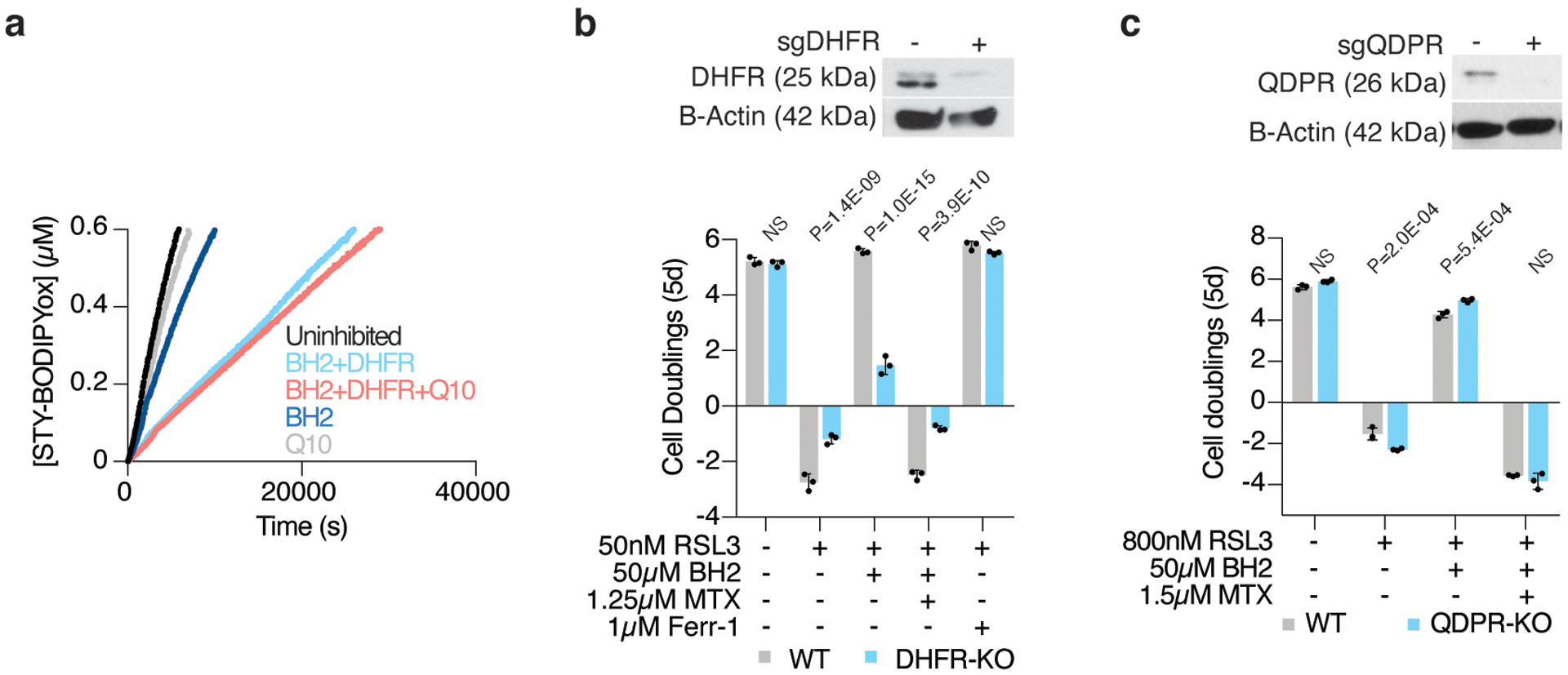
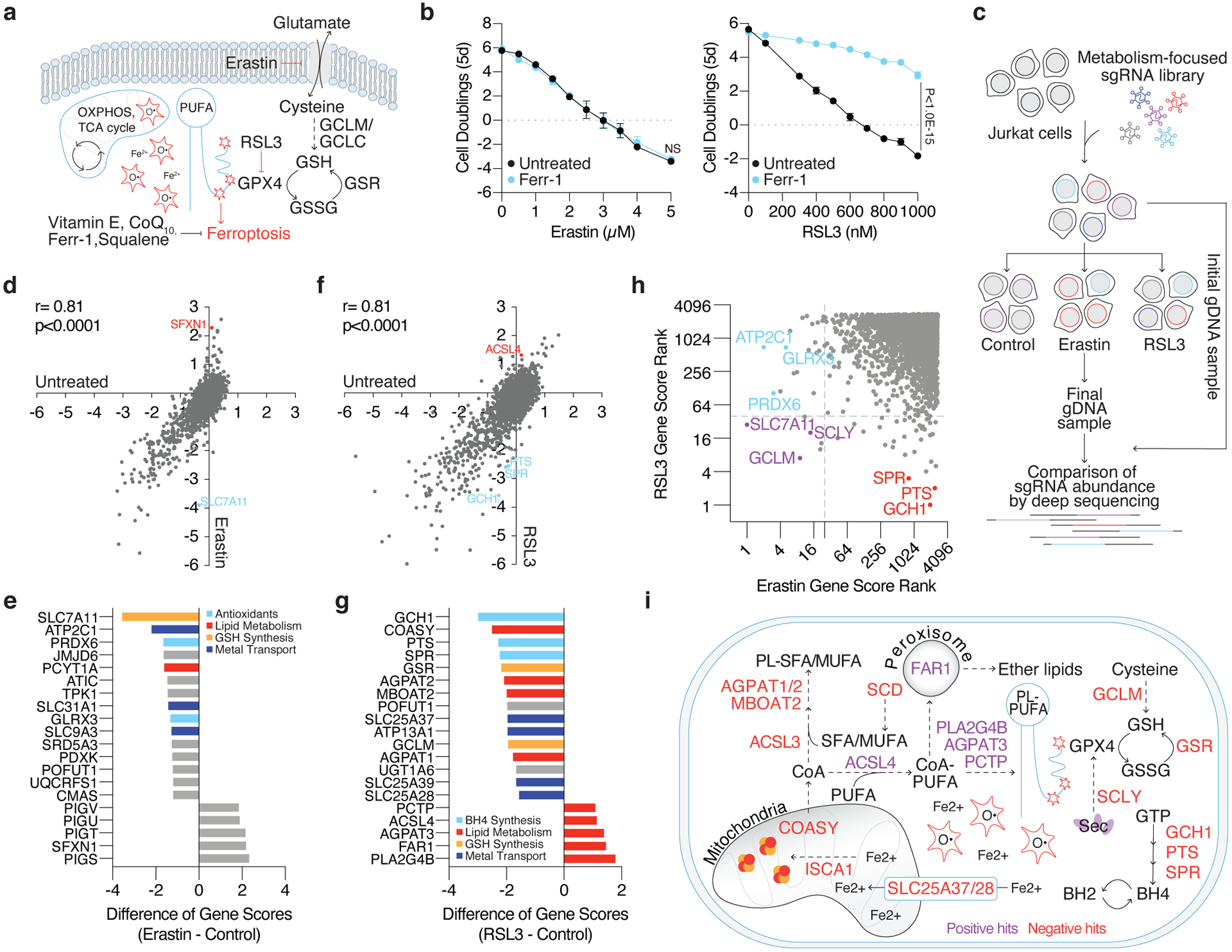
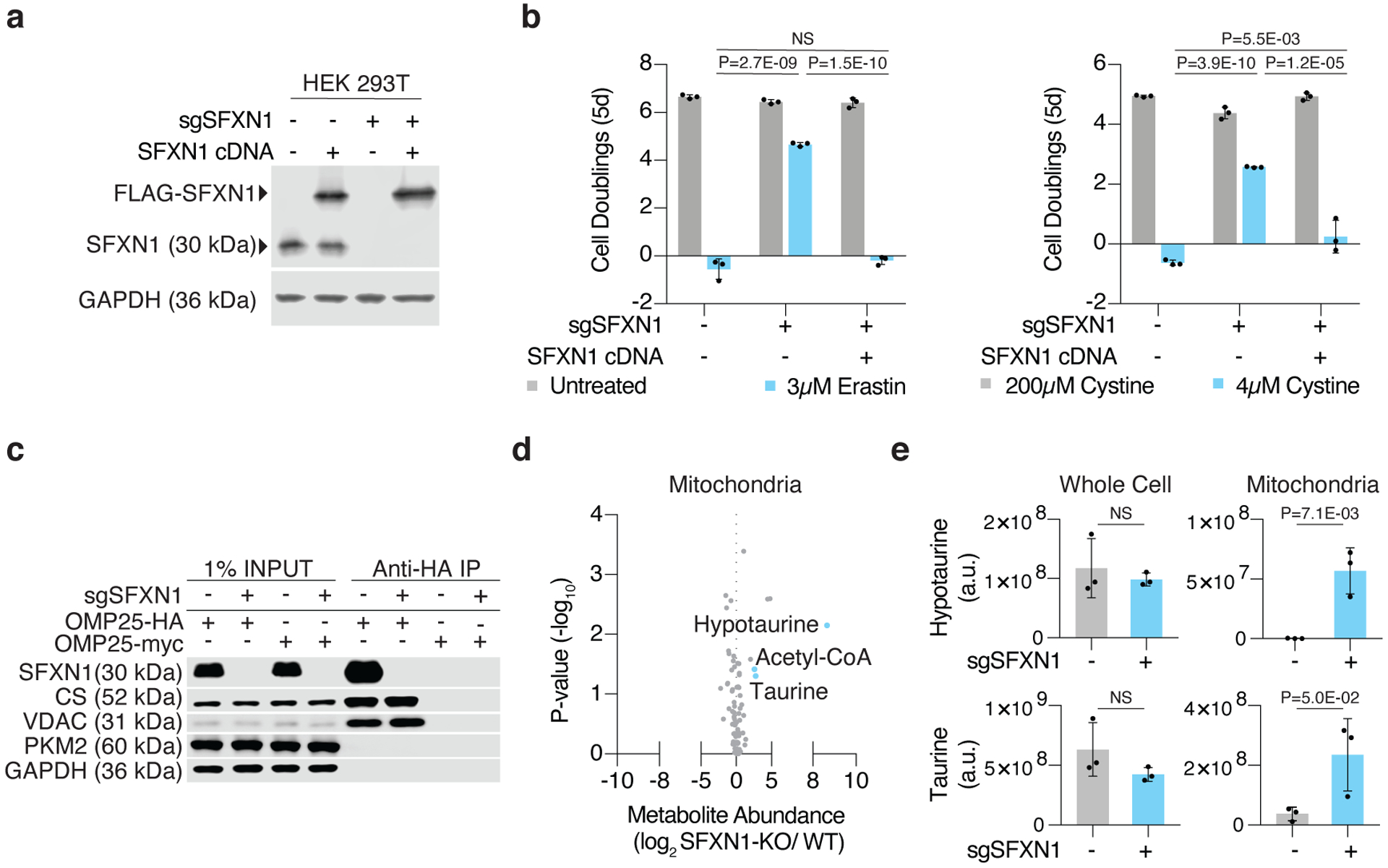
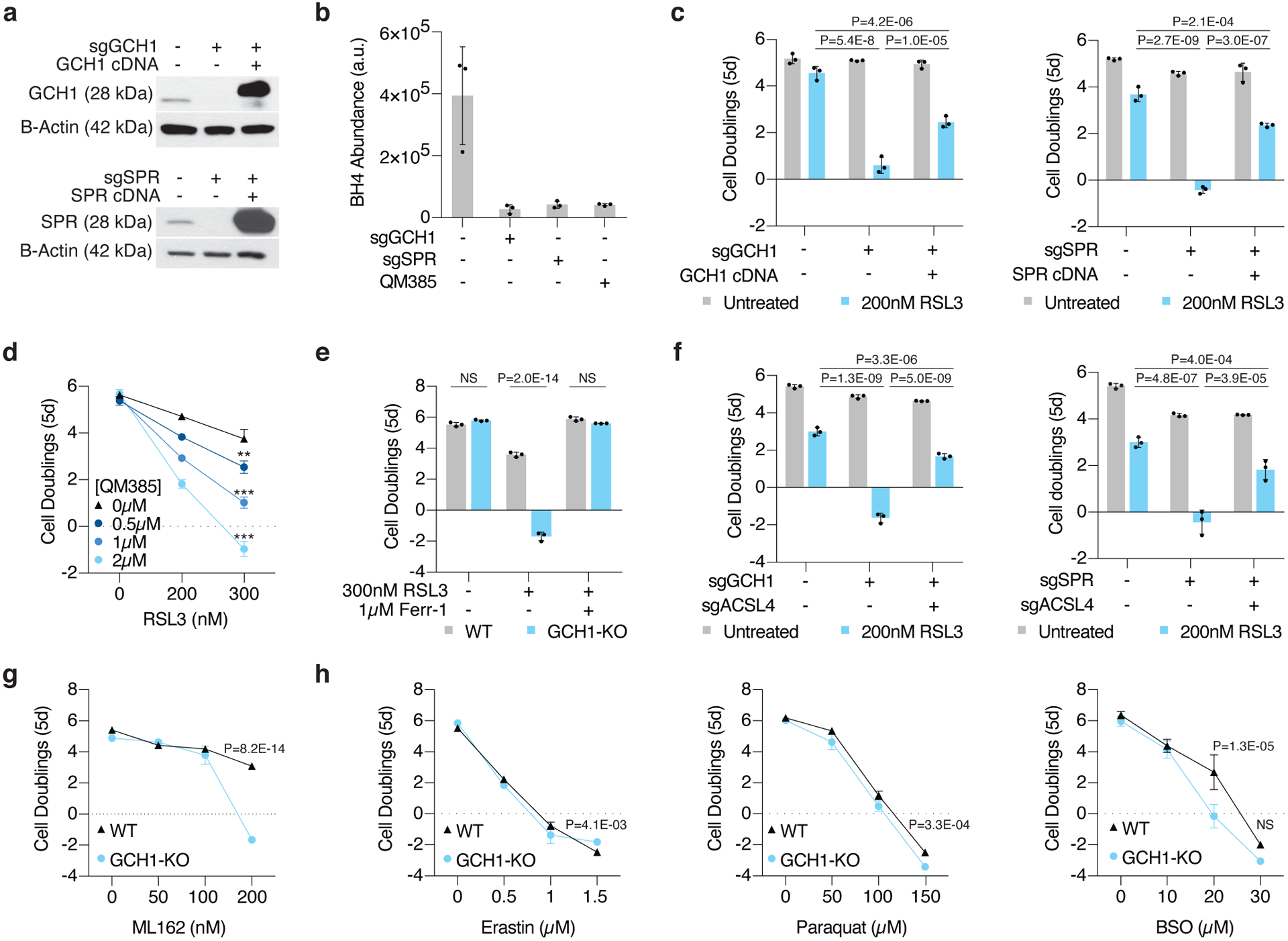
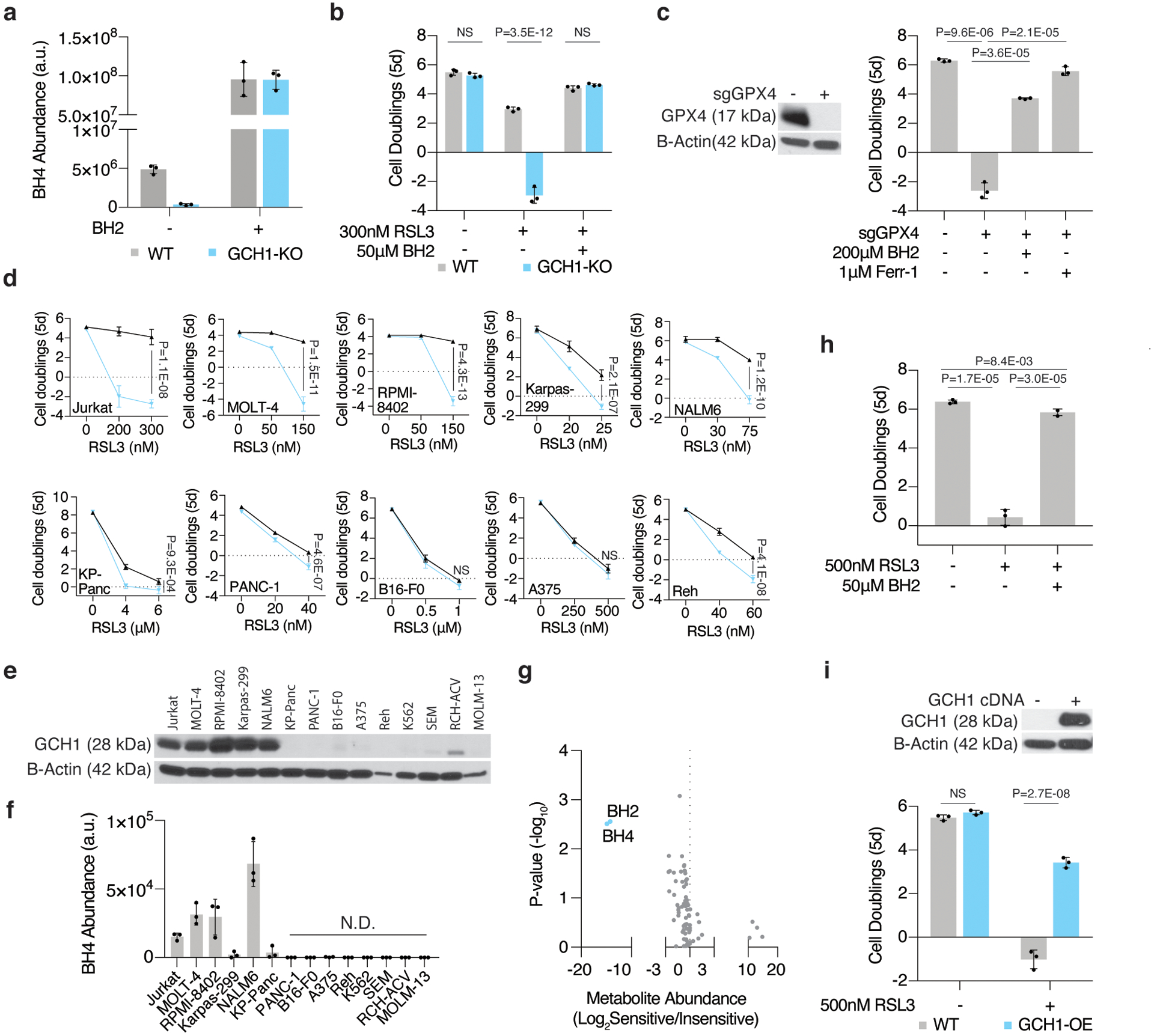
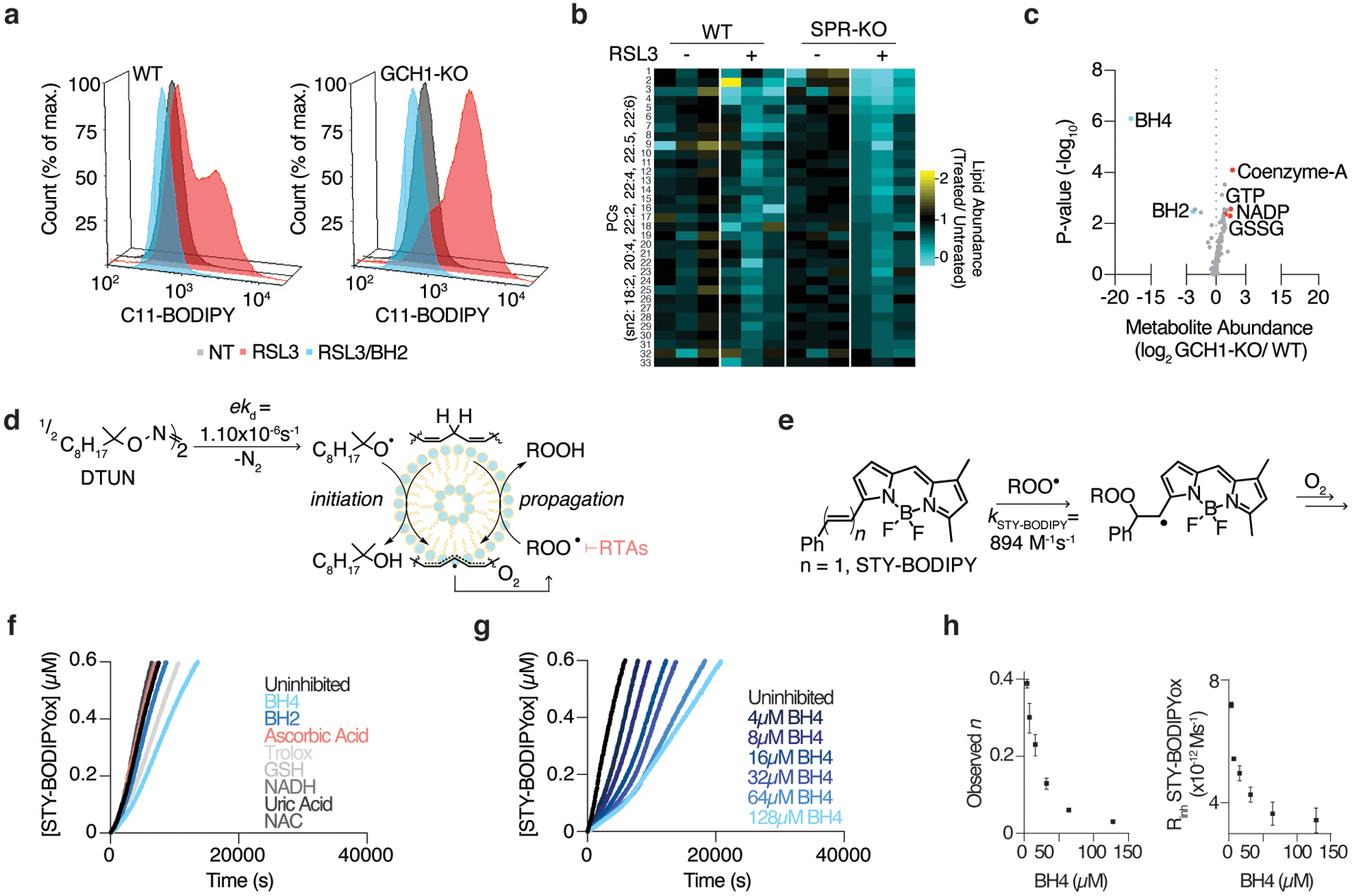
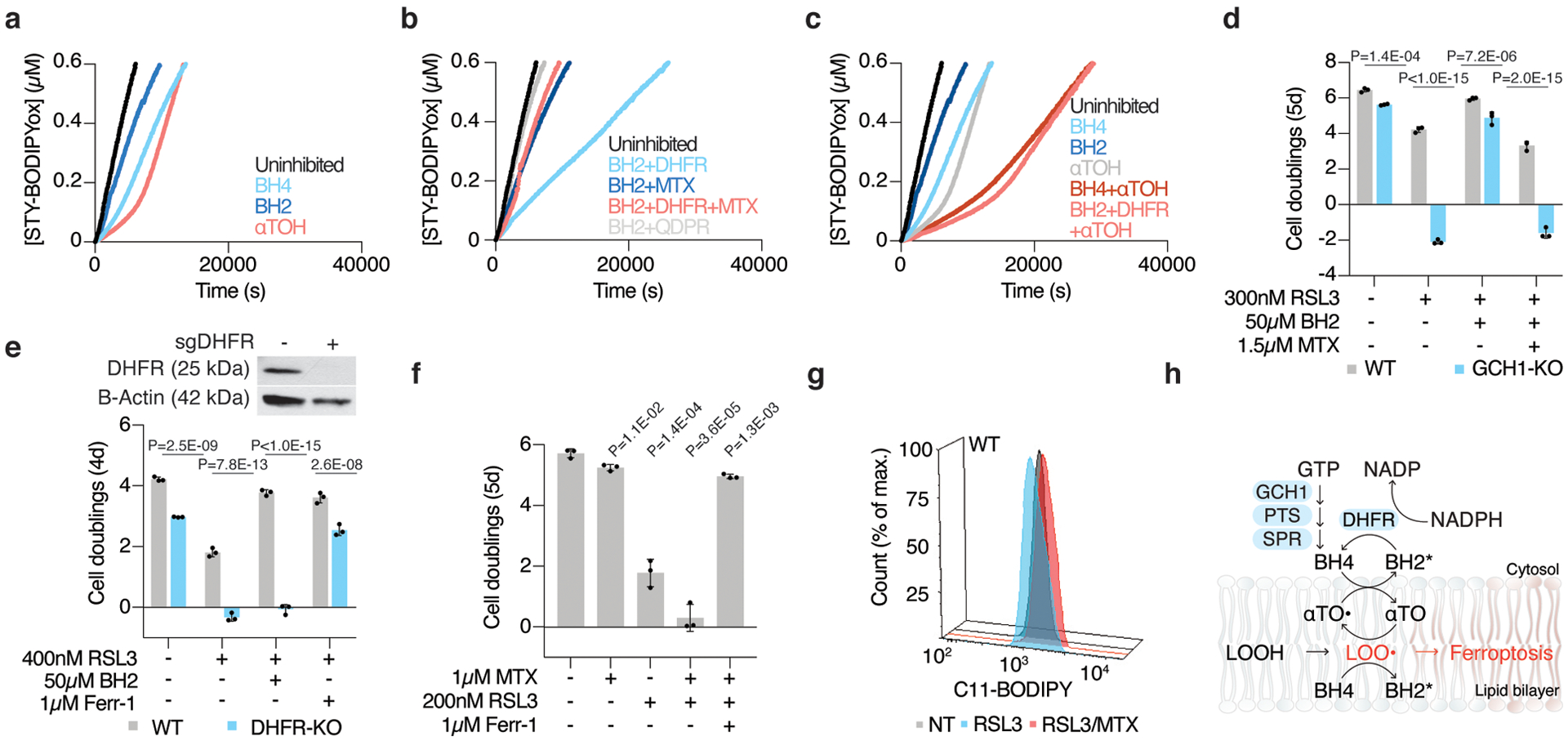
Cancer cells rewire their metabolism and rely on endogenous antioxidants to mitigate lethal oxidative damage to lipids. However, the metabolic processes that modulate the response to lipid peroxidation are poorly defined. Using genetic screens, we compared metabolic genes essential for proliferation upon inhibition of cystine uptake or glutathione peroxidase-4 (GPX4). Interestingly, very few genes were commonly required under both conditions, suggesting that cystine limitation and GPX4 inhibition may impair proliferation via distinct mechanisms. Our screens also identify tetrahydrobiopterin (BH4) biosynthesis as an essential metabolic pathway upon GPX4 inhibition. Mechanistically, BH4 is a potent radical-trapping antioxidant that protects lipid membranes from autoxidation, alone and in synergy with vitamin E. Dihydrofolate reductase catalyzes the regeneration of BH4, and its inhibition by methotrexate synergizes with GPX4 inhibition. Altogether, our work identifies the mechanism by which BH4 acts as an endogenous antioxidant and provides a compendium of metabolic modifiers of lipid peroxidation.
Figures
References
-
- Sies H, Berndt C & Jones DP Oxidative Stress. Annu. Rev. Biochem 86, 715–748 (2017). - PubMed
-
- Yin H, Xu L & Porter NA Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev 111, 5944–5972 (2011). - PubMed
-
- Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019). - PubMed
-
- Ingold KU & Pratt DA Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev 114, 9022–9046 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
