

Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death
- PMID: 32780352
- PMCID: PMC7530023
- DOI: 10.1007/s12035-020-02034-w
Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death
Abstract
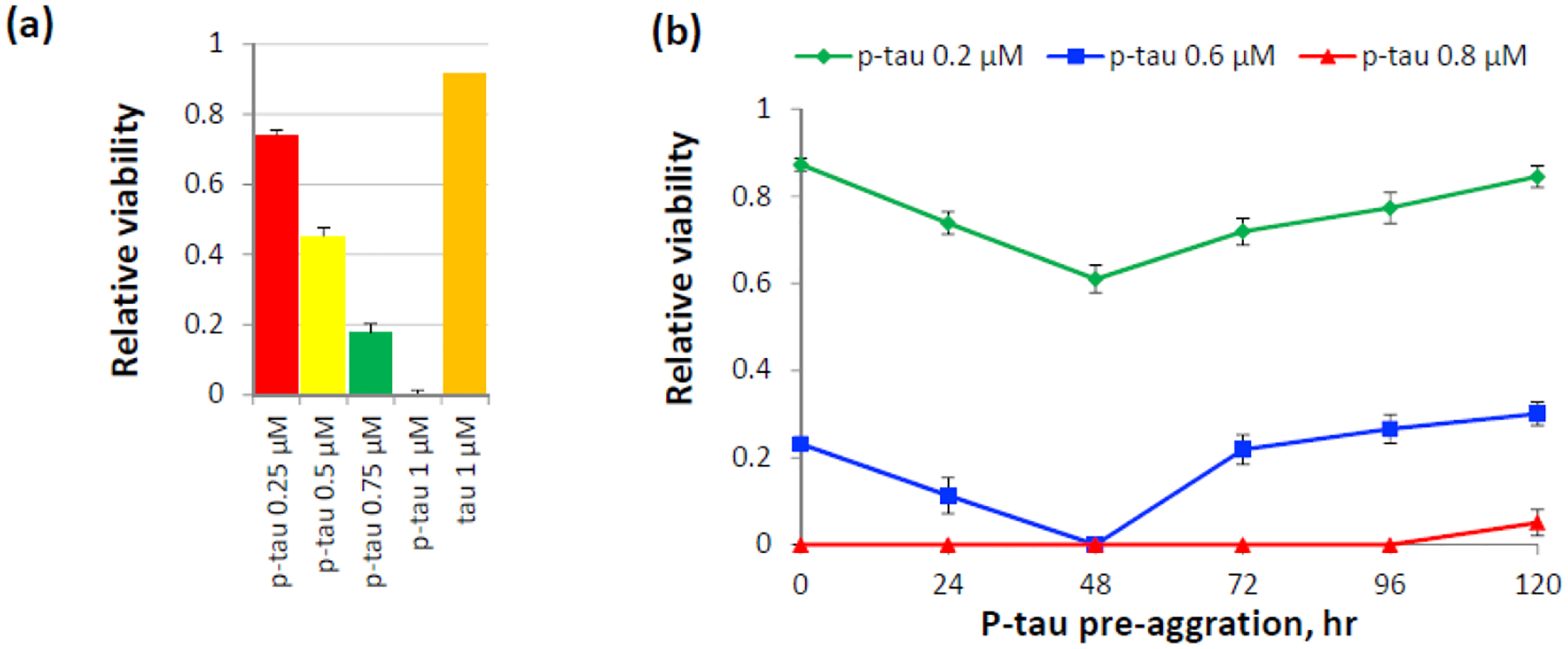
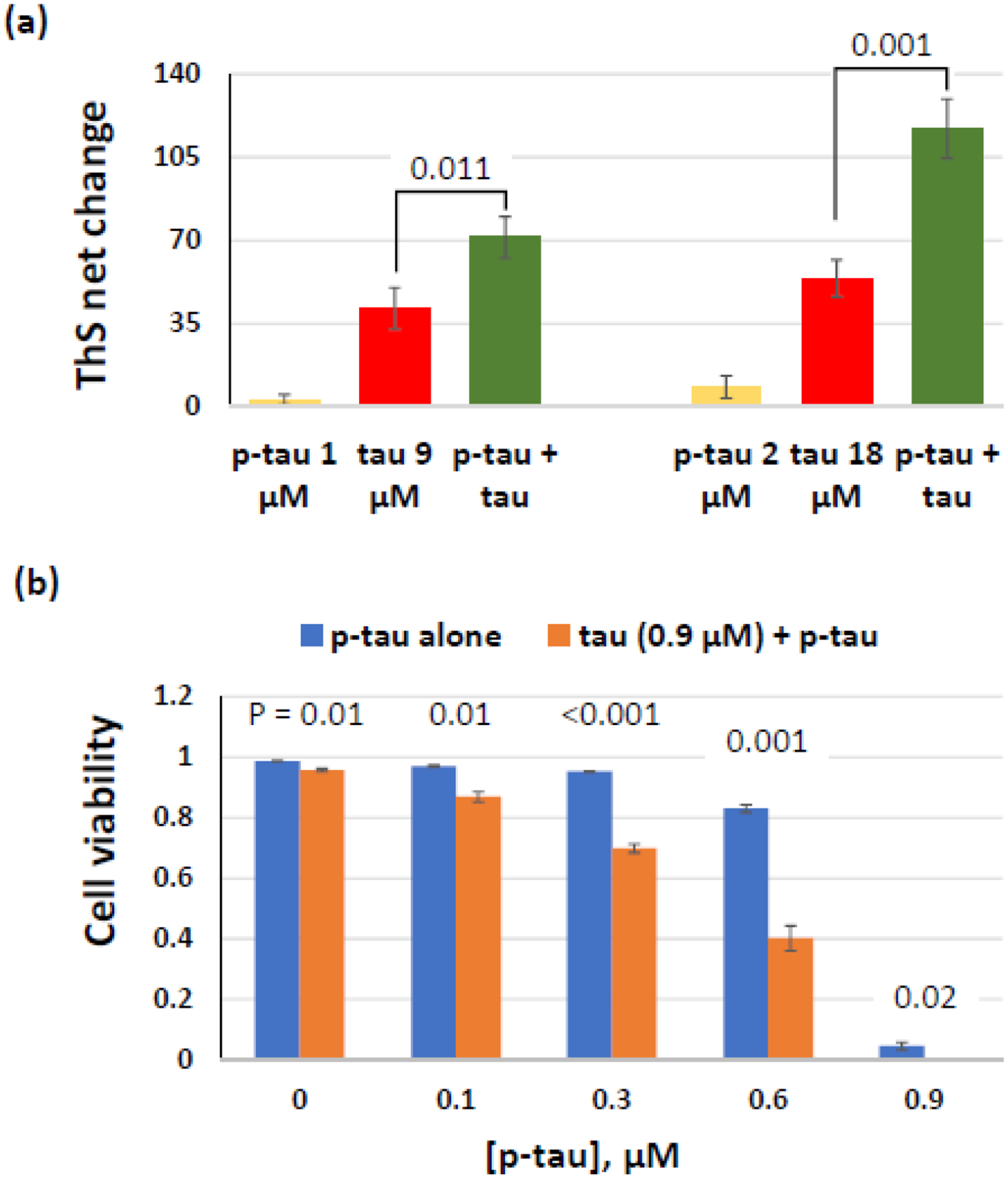
Alzheimer's disease (AD) is a neurodegenerative disorder without a cure or prevention to date. Hyperphosphorylated tau forms the neurofibrillary tangles (NFTs) that correlate well with the progression of cognitive impairments. Animal studies demonstrated the pathogenic role of hyperphosphorylated tau. Understanding how abnormal phosphorylation renders a normal tau prone to form toxic fibrils is key to delineating molecular pathology and to developing efficacious drugs for AD. Production of a tau bearing the disease-relevant hyperphosphorylation and molecular characters is a pivotal step. Here, we report the preparation and characterization of a recombinant hyperphosphorylated tau (p-tau) with strong relevance to disease. P-tau generated by the PIMAX approach resulted in phosphorylation at multiple epitopes linked to the progression of AD neuropathology. In stark contrast to unmodified tau that required an aggregation inducer, and which had minimal effects on cell functions, p-tau formed inducer-free fibrils that triggered a spike of mitochondrial superoxide, induced apoptosis, and caused cell death at sub-micromolar concentrations. P-tau-induced apoptosis was suppressed by inhibitors for reactive oxygen species. Hyperphosphorylation apparently caused rapid formation of a disease-related conformation. In both aggregation and cytotoxicity, p-tau exhibited seeding activities that converted the unmodified tau into a cytotoxic species with an increased propensity for fibrillization. These characters of p-tau are consistent with the emerging view that hyperphosphorylation causes tau to become an aggregation-prone and cytotoxic species that underlies diffusible pathology in AD and other tauopathies. Our results further suggest that p-tau affords a feasible tool for Alzheimer's disease mechanistic and drug discovery studies.
Keywords: Alzheimer’s disease; Hyperphosphorylated tau; Neurofibrillary tangle; Tauopathy.
Conflict of interest statement
Figures




References
-
- Glenner GG & Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochem Biophys Res Commun. 120, 885–90. - PubMed
-
- Hardy J & Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics, Science. 297, 353–6. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
