

Deciphering Cell Fate Decision by Integrated Single-Cell Sequencing Analysis
- PMID: 32780577
- PMCID: PMC7115822
- DOI: 10.1146/annurev-biodatasci-111419-091750
Deciphering Cell Fate Decision by Integrated Single-Cell Sequencing Analysis
Abstract
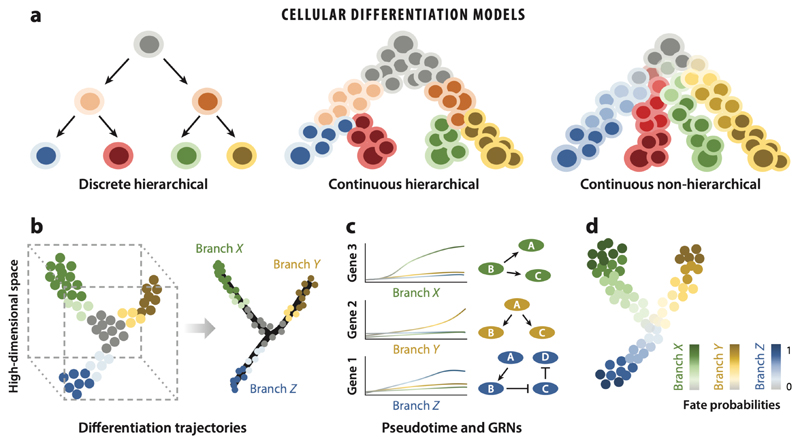
Cellular differentiation is a common underlying feature of all multicellular organisms through which naïve cells progressively become fate restricted and develop into mature cells with specialized functions. A comprehensive understanding of the regulatory mechanisms of cell fate choices during de- velopment, regeneration, homeostasis, and disease is a central goal of mod- ern biology. Ongoing rapid advances in single-cell biology are enabling the exploration of cell fate specification at unprecedented resolution. Here, we review single-cell RNA sequencing and sequencing of other modalities as methods to elucidate the molecular underpinnings of lineage specification. We specifically discuss how the computational tools available to reconstruct lineage trajectories, quantify cell fate bias, and perform dimensionality re- duction for data visualization are providing new mechanistic insights into the process of cell fate decision. Studying cellular differentiation using single- cell genomic tools is paving the way for a detailed understanding of cellular behavior in health and disease.
Conflict of interest statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Figures


References
-
- Kopp JL, Grompe M, Sander M. Stem cells versus plasticity in liver and pancreas regeneration. Nat Cell Biol. 2016;18:238–45. - PubMed
-
- Brockes JP, Kumar A. Appendage regeneration in adult vertebrates and implications for regenerative medicine. Science. 2005;310:1919–23. - PubMed
-
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–98. - PubMed
Grants and funding
- GR4980/1-1/Max Planck Society, the Ger- man Research Foundation (DFG)/International
- GRK2344/Max Planck Society, the Ger- man Research Foundation (DFG)/International
- 818846/ERC_/European Research Council/International
- GR4980/3-1/Max Planck Society, the Ger- man Research Foundation (DFG)/International
- EXC-2189/Germany’s Excellence Strategy (Centre for Integrative Biology Sig- nalling Studies grant EXC-2189, project ID 390939984)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
