

Anticoagulant and signaling functions of antithrombin
- PMID: 32780936
- PMCID: PMC7855051
- DOI: 10.1111/jth.15052
Anticoagulant and signaling functions of antithrombin
Abstract
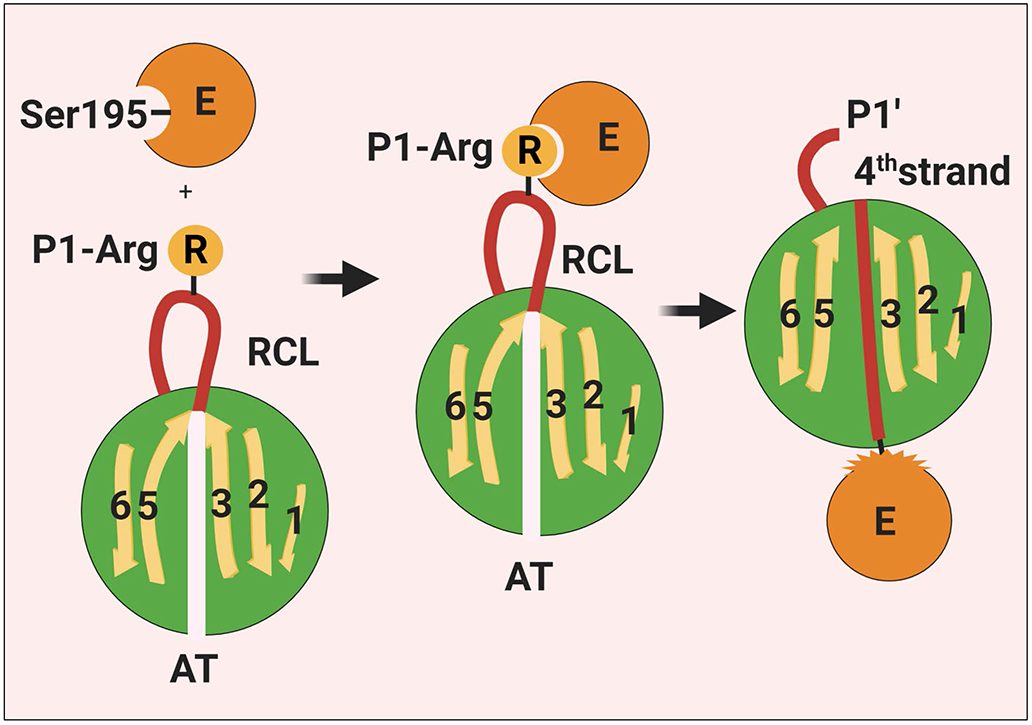
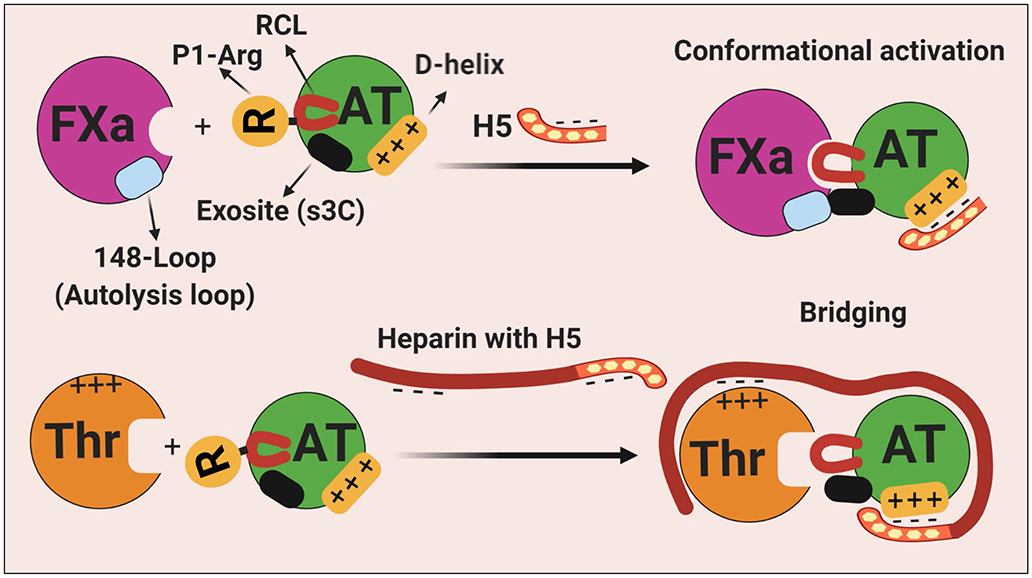
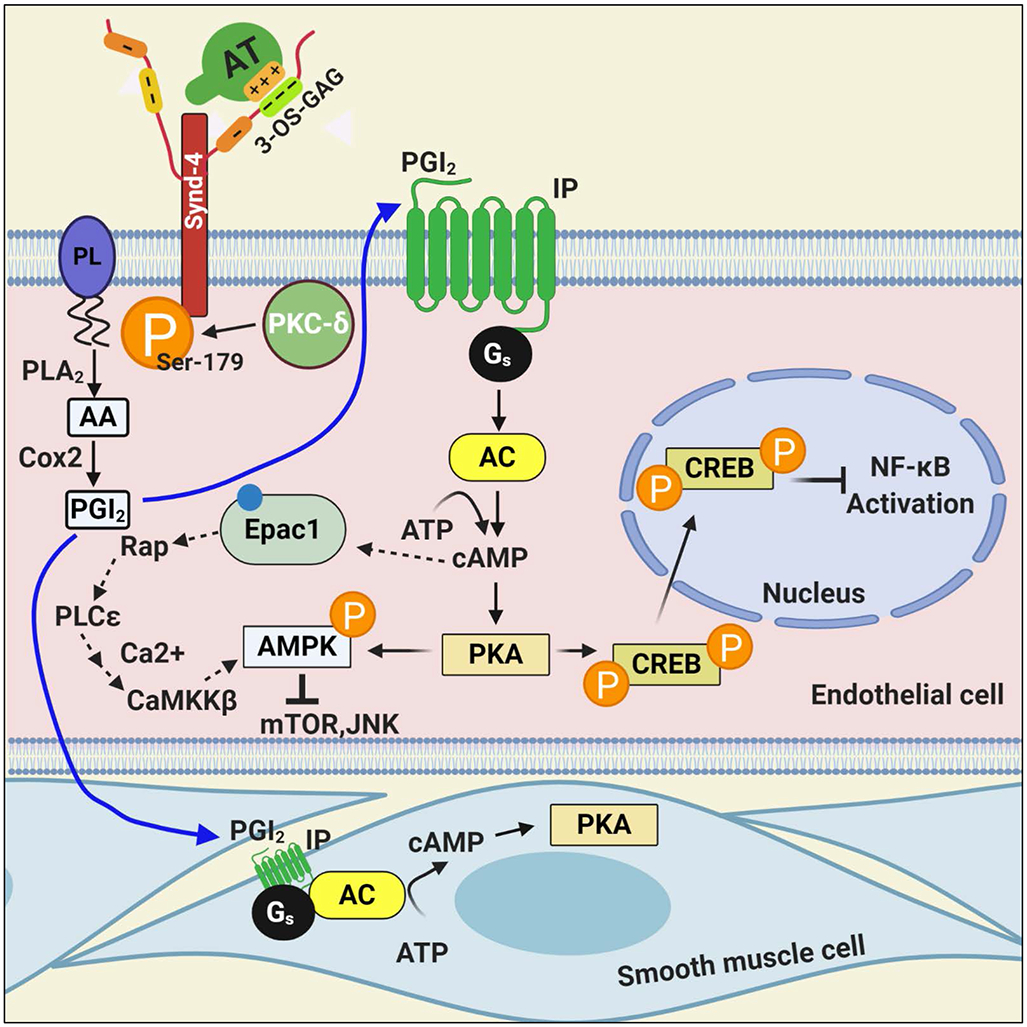
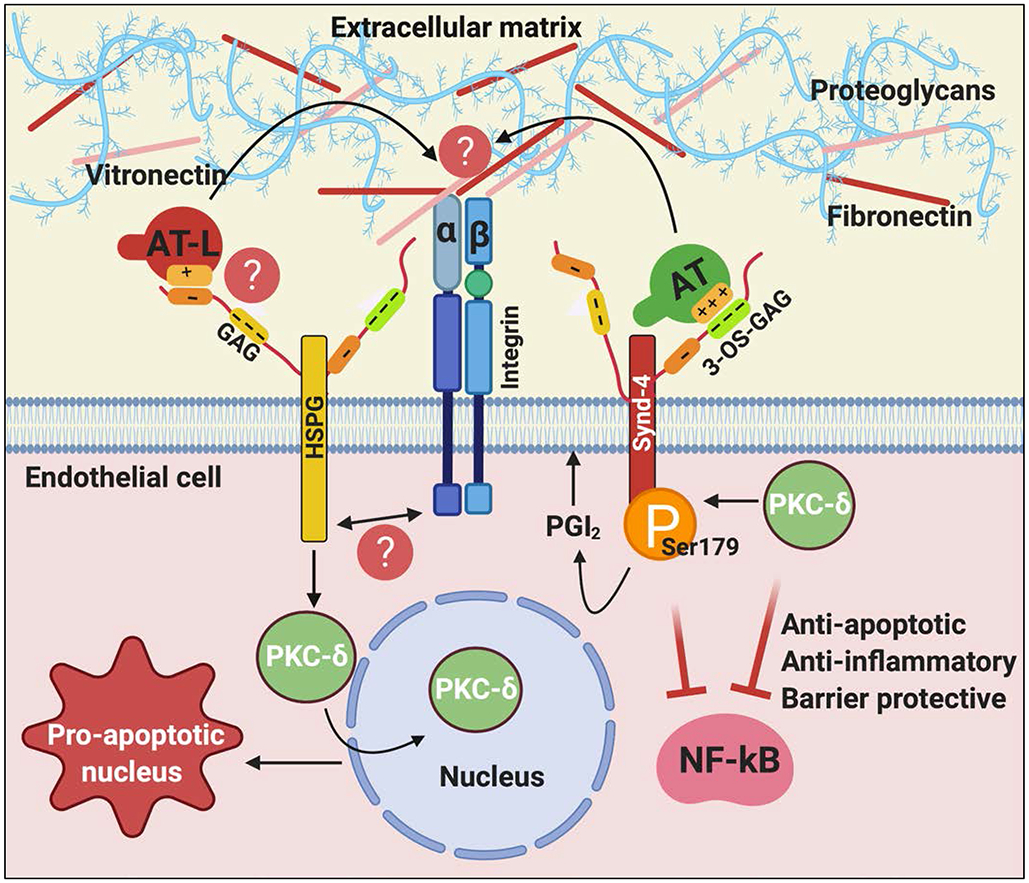
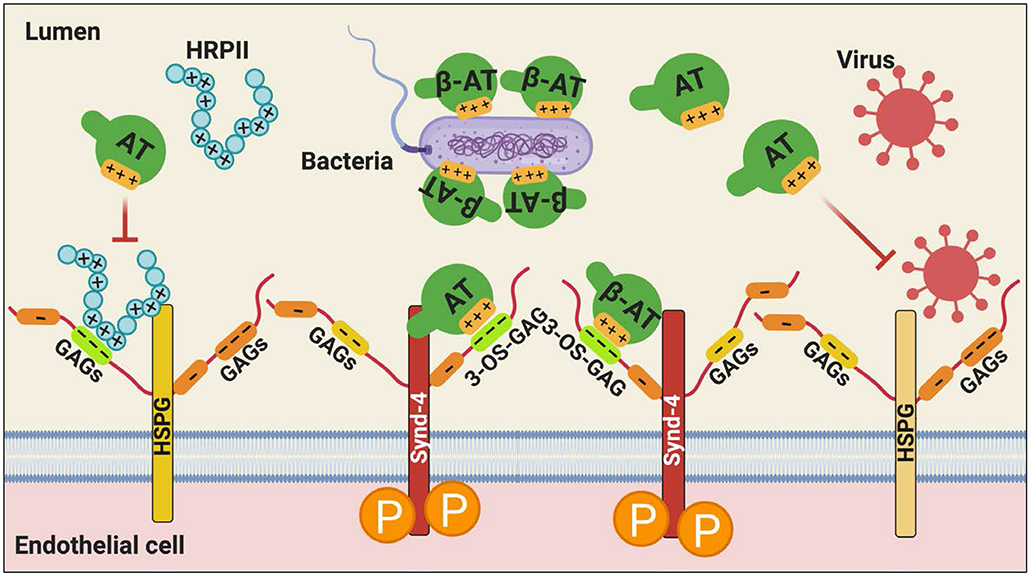
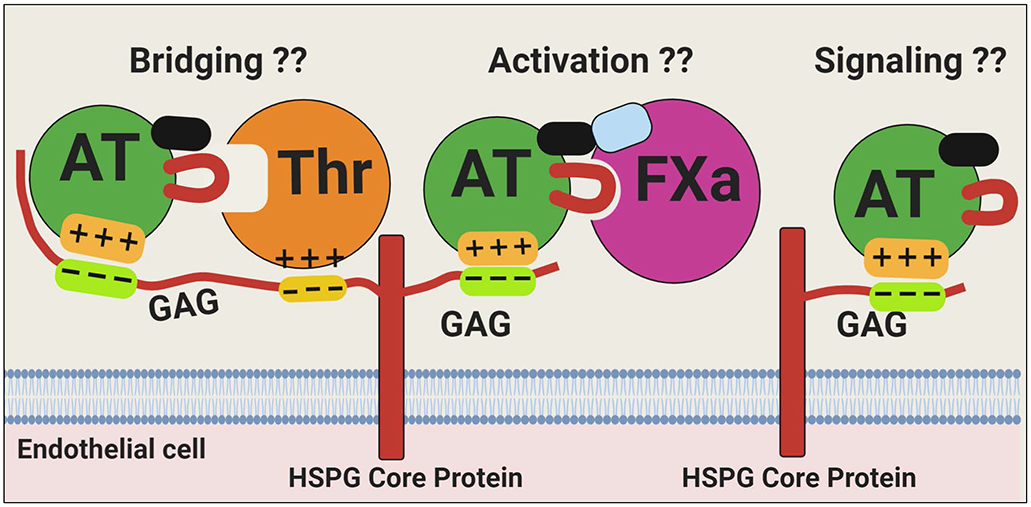
Antithrombin (AT) is a major plasma glycoprotein of the serpin superfamily that regulates the proteolytic activity of the procoagulant proteases of both intrinsic and extrinsic pathways. Two important structural features that participate in the regulatory function of AT include a mobile reactive center loop that binds to active site of coagulation proteases, trapping them in the form of inactive covalent complexes, and a basic D-helix that binds to therapeutic heparins and heparan sulfate proteoglycans (HSPGs) on vascular endothelial cells. The binding of D-helix of AT by therapeutic heparins promotes the reactivity of the serpin with coagulation proteases by several orders of magnitude by both a conformational activation of the serpin and a template (bridging) mechanism. In addition to its essential anticoagulant function, AT elicits a potent anti-inflammatory signaling response when it binds to distinct vascular endothelial cell HSPGs, thereby inducing prostacyclin synthesis. Syndecans-4 has been found as a specific membrane-bound HSPG receptor on endothelial cells that relays the signaling effect of AT to the relevant second messenger molecules in the signal transduction pathways inside the cell. However, following cleavage by coagulation proteases and/or by spontaneous conversion to a latent form, AT loses both its anti-inflammatory activity and high-affinity interaction with heparin and HSPGs. Interestingly, these low-affinity heparin conformers of AT elicit potent proapoptotic and antiangiogenic activities by also binding to specific HSPGs by unknown mechanisms. This review article will summarize current knowledge about mechanisms through which different conformers of AT exert their serine protease inhibitory and intracellular signaling functions in these biological pathways.
Keywords: anti-inflammatory; anticoagulant; antithrombin; heparan sulfate; heparin.
© 2020 International Society on Thrombosis and Haemostasis.
Conflict of interest statement
Disclosure of Conflict of Interests
The authors declare no conflict of interests.
Figures
References
-
- Gettins PG. Serpins structure, mechanism, and function. Chem Rev. 2002; 102: 4751–4803. - PubMed
-
- Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973; 246: 355–357. - PubMed
-
- Carrell RW, Skinner R, Jin L, Abrahams JP. Structural Mobility of Antithrombin and its Modulation by Heparin. Thromb Haemost. 1997; 78: 516–519. - PubMed
-
- Leon M, Aiach M, Coezy E, Gunennec JY, Feissinger JN. Antithrombin III synthesis in rat liver parenchymal cells. Thromb Res. 1983; 30: 369–375. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
