

Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer
- PMID: 32782288
- PMCID: PMC7419503
- DOI: 10.1038/s41598-020-70449-5
Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer
Abstract
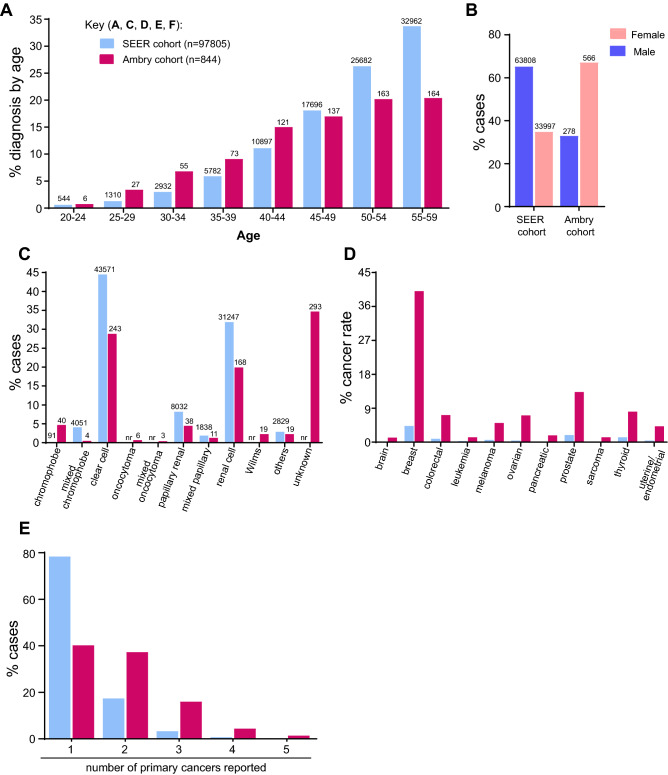
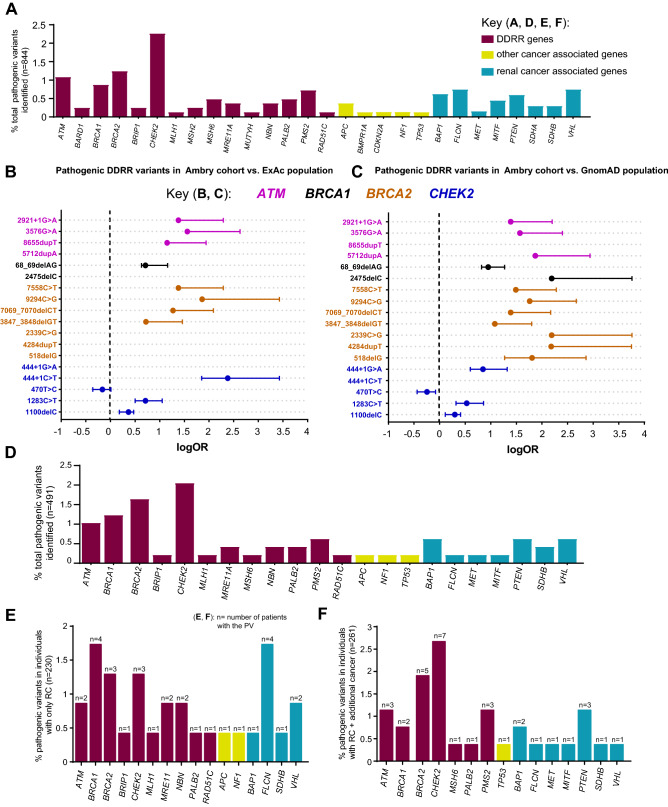
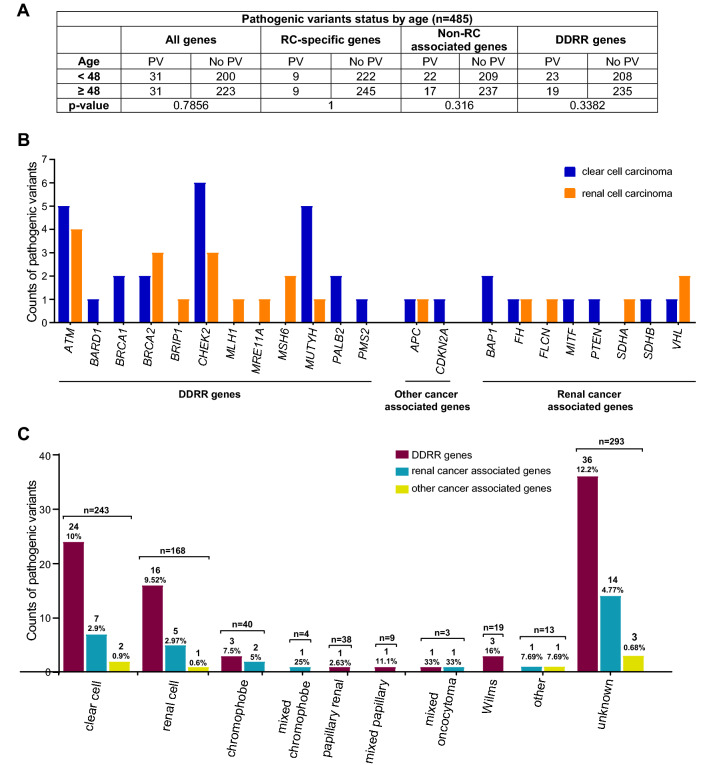
Pathogenic variants (PVs) in multiple genes are known to increase the risk of early-onset renal cancer (eoRC). However, many eoRC patients lack PVs in RC-specific genes; thus, their genetic risk remains undefined. Here, we determine if PVs in DNA damage response and repair (DDRR) genes are enriched in eoRC patients undergoing cancer risk assessment. Retrospective review of de-identified results from 844 eoRC patients, undergoing testing with a multi-gene panel, for a variety of indications, by Ambry Genetics. PVs in cancer-risk genes were identified in 12.8% of patients-with 3.7% in RC-specific, and 8.55% in DDRR genes. DDRR gene PVs were most commonly identified in CHEK2, BRCA1, BRCA2, and ATM. Among the 2.1% of patients with a BRCA1 or BRCA2 PV, < 50% reported a personal history of hereditary breast or ovarian-associated cancer. No association between age of RC diagnosis and prevalence of PVs in RC-specific or DDRR genes was observed. Additionally, 57.9% patients reported at least one additional cancer; breast cancer being the most common (40.1% of females, 2.5% of males). Multi-gene testing including DDRR genes may provide a more comprehensive risk assessment in eoRC patients. Further validation is needed to characterize the association with eoRC.
Conflict of interest statement
A.F. has consulted for Invitae Corporation, and received speaker fees from AstraZeneca, M.J.H. performs collaborative research (with no funding) with the following: Myriad Genetics, Invitae Corporation, Ambry Genetics, Foundation Medicine, Inc. He also performs collaborative research (with no funding) and is part of a Precision Oncology Alliance funded by Caris Life Sciences (cover travel and meals at meetings), L.H., M.R. and V.S. are full-time salaried employees of Ambry Genetics. All other authors report no conflicts or disclosures.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
