

Gut microbial co-abundance networks show specificity in inflammatory bowel disease and obesity
- PMID: 32782301
- PMCID: PMC7419557
- DOI: 10.1038/s41467-020-17840-y
Gut microbial co-abundance networks show specificity in inflammatory bowel disease and obesity
Abstract
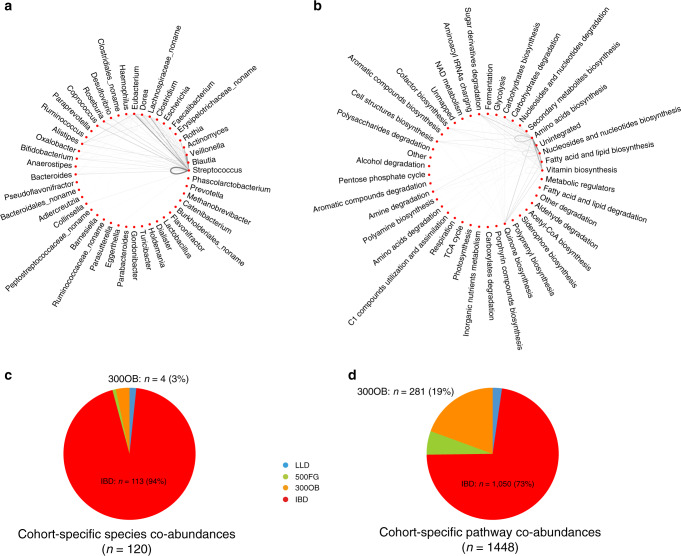
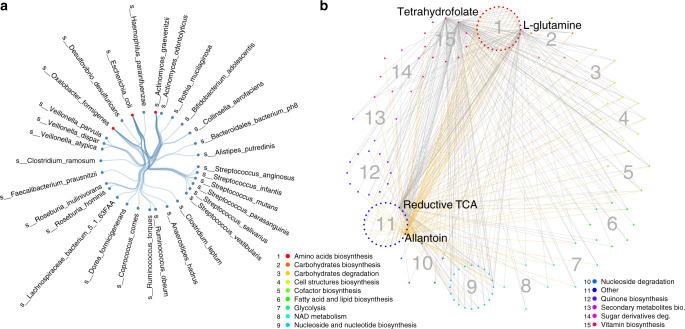
The gut microbiome is an ecosystem that involves complex interactions. Currently, our knowledge about the role of the gut microbiome in health and disease relies mainly on differential microbial abundance, and little is known about the role of microbial interactions in the context of human disease. Here, we construct and compare microbial co-abundance networks using 2,379 metagenomes from four human cohorts: an inflammatory bowel disease (IBD) cohort, an obese cohort and two population-based cohorts. We find that the strengths of 38.6% of species co-abundances and 64.3% of pathway co-abundances vary significantly between cohorts, with 113 species and 1,050 pathway co-abundances showing IBD-specific effects and 281 pathway co-abundances showing obesity-specific effects. We can also replicate these IBD microbial co-abundances in longitudinal data from the IBD cohort of the integrative human microbiome (iHMP-IBD) project. Our study identifies several key species and pathways in IBD and obesity and provides evidence that altered microbial abundances in disease can influence their co-abundance relationship, which expands our current knowledge regarding microbial dysbiosis in disease.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Chen LM, Garmaeva S, Zhernakova A, Fu JY, Wijmenga C. A system biology perspective on environment-host-microbe interactions. Hum. Mol. Genet. 2018;27:R187–R194. - PubMed
-
- Falony G, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–564. - PubMed
-
- Kurilshikov A, Wijmenga C, Fu J, Zhernakova A. Host genetics and gut microbiome: challenges and perspectives. Trends Immunol. 2017;38:633–647. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
