

Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study
- PMID: 32784459
- PMCID: PMC7460976
- DOI: 10.3390/ijms21165688
Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study
Abstract
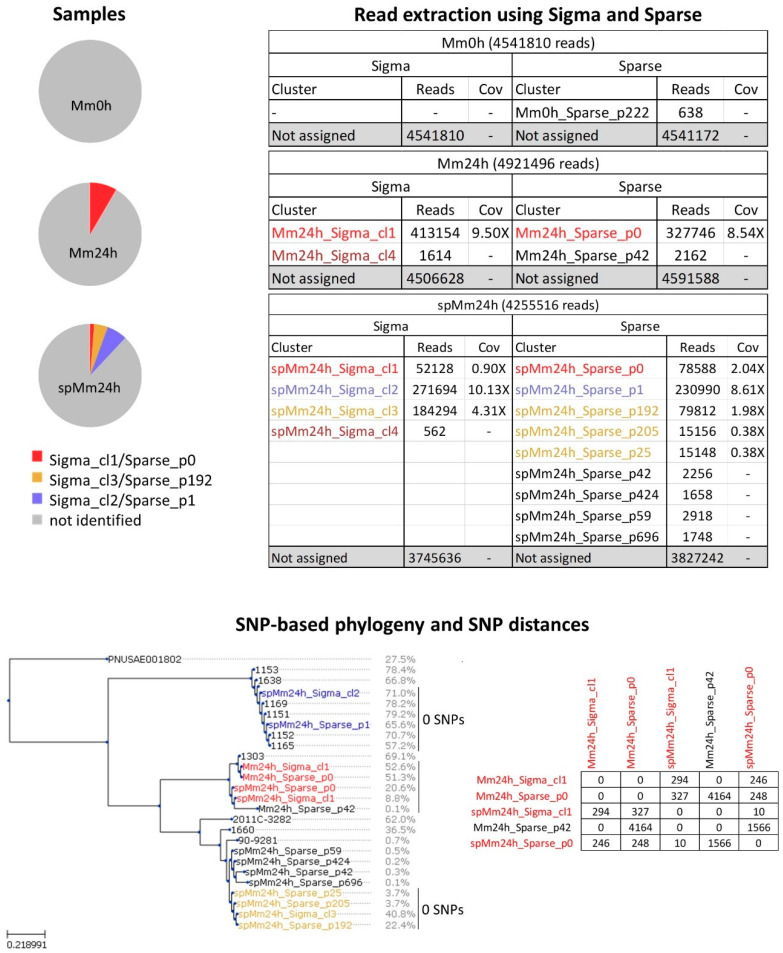
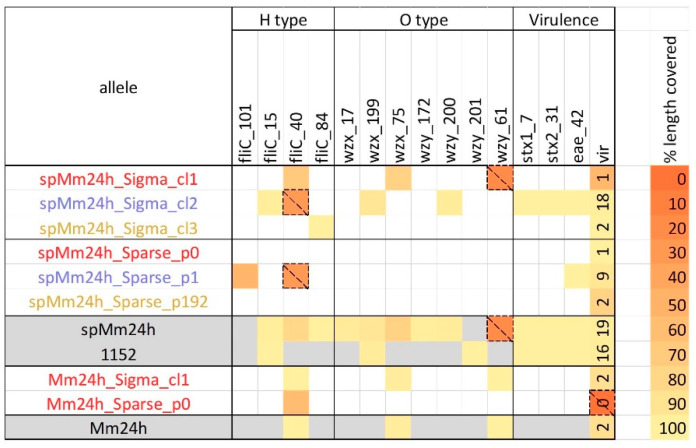
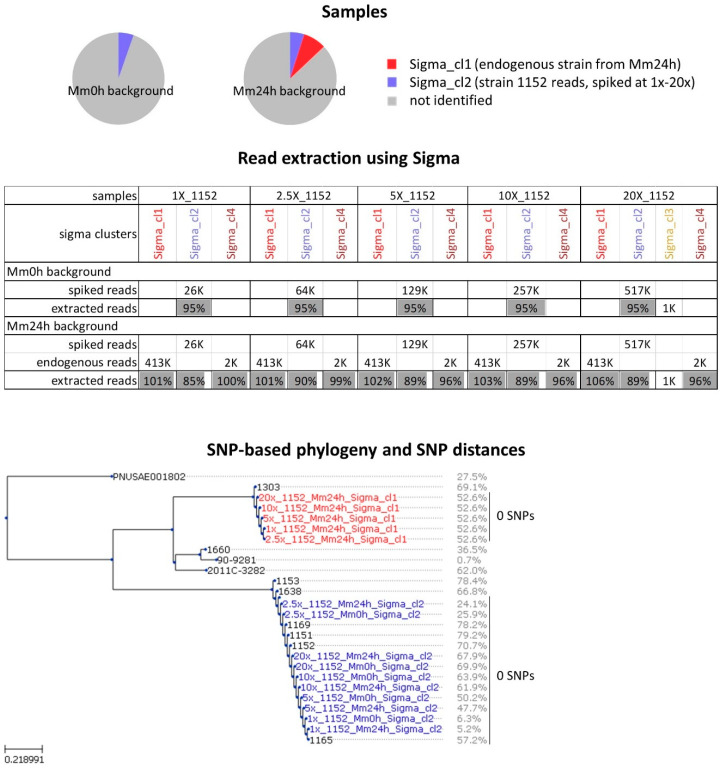
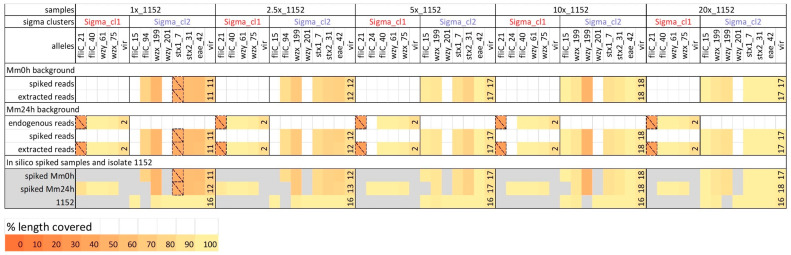
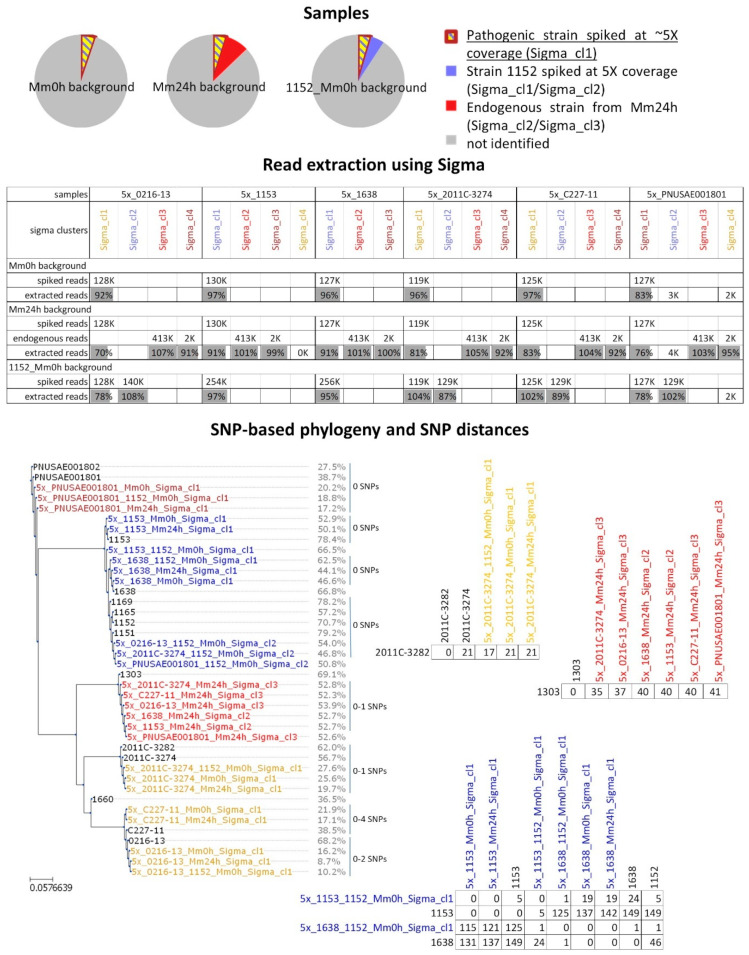
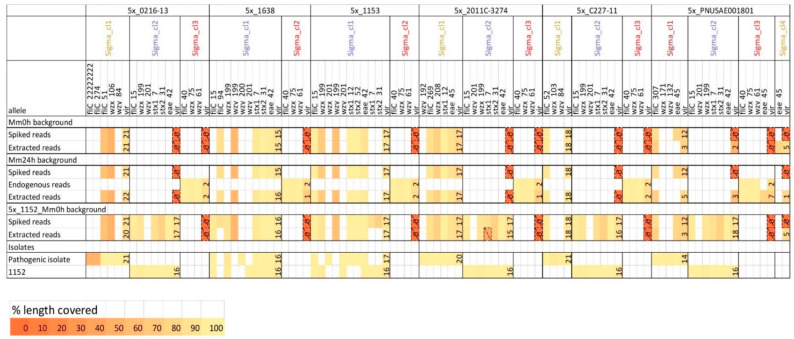
Culture-independent diagnostics, such as metagenomic shotgun sequencing of food samples, could not only reduce the turnaround time of samples in an outbreak investigation, but also allow the detection of multi-species and multi-strain outbreaks. For successful foodborne outbreak investigation using a metagenomic approach, it is, however, necessary to bioinformatically separate the genomes of individual strains, including strains belonging to the same species, present in a microbial community, which has up until now not been demonstrated for this application. The current work shows the feasibility of strain-level metagenomics of enriched food matrix samples making use of data analysis tools that classify reads against a sequence database. It includes a brief comparison of two database-based read classification tools, Sigma and Sparse, using a mock community obtained by in vitro spiking minced meat with a Shiga toxin-producing Escherichia coli (STEC) isolate originating from a described outbreak. The more optimal tool Sigma was further evaluated using in silico simulated metagenomic data to explore the possibilities and limitations of this data analysis approach. The performed analysis allowed us to link the pathogenic strains from food samples to human isolates previously collected during the same outbreak, demonstrating that the metagenomic approach could be applied for the rapid source tracking of foodborne outbreaks. To our knowledge, this is the first study demonstrating a data analysis approach for detailed characterization and phylogenetic placement of multiple bacterial strains of one species from shotgun metagenomic WGS data of an enriched food sample.
Keywords: foodborne outbreak investigation; public health; strain-level metagenomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Leopold S.R., Goering R.V., Witten A., Harmsen D., Mellmann A. Bacterial whole-genome sequencing revisited: Portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 2014;52:2365–2370. doi: 10.1128/JCM.00262-14. - DOI - PMC - PubMed
-
- EFSA BIOHAZ Panel. Koutsoumanis K., Allende A., Alvarez-Ordóñez A., Bolton D., Bover-Cid S., Chemaly M., Davies R., De Cesare A., Hilbert F. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA. J. 2019;17:e05898. - PMC - PubMed
-
- Rantsiou K., Kathariou S., Winkler A., Skandamis P., Saint-Cyr M.J., Rouzeau-Szynalski K., Amézquita A. Next generation microbiological risk assessment: Opportunities of whole genome sequencing (WGS) for foodborne pathogen surveillance, source tracking and risk assessment. Int. J. Food Microbiol. 2018;287:3–9. doi: 10.1016/j.ijfoodmicro.2017.11.007. - DOI - PubMed
-
- World Health Organization Whole Genome Sequencing for Foodborne Disease Surveillance: Landscape Paper. [(accessed on 1 July 2020)]; Available online: http://origin.who.int/foodsafety/publications/foodborne_disease/wgs_land...
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
