

Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies
- PMID: 32785274
- PMCID: PMC7423102
- DOI: 10.1371/journal.pone.0237300
Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies
Abstract
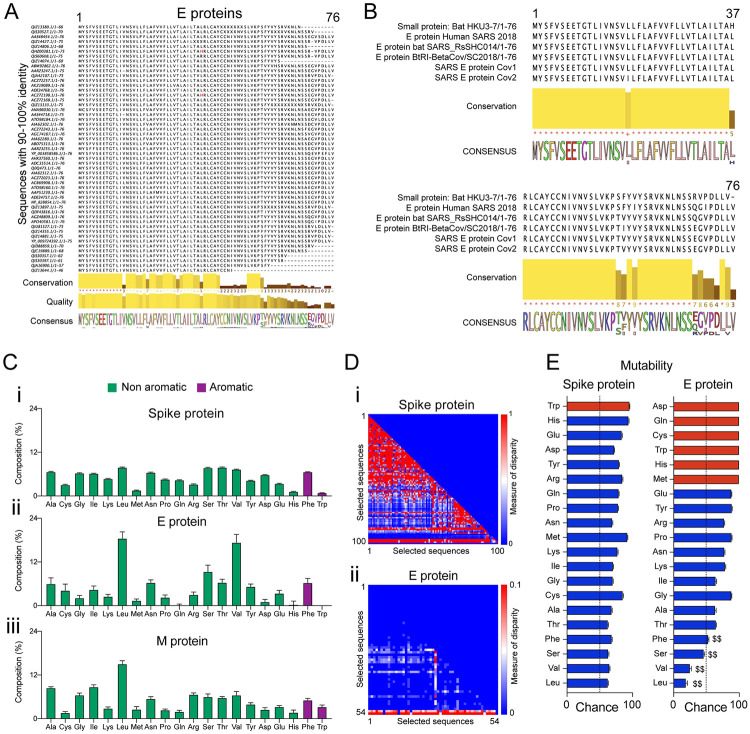
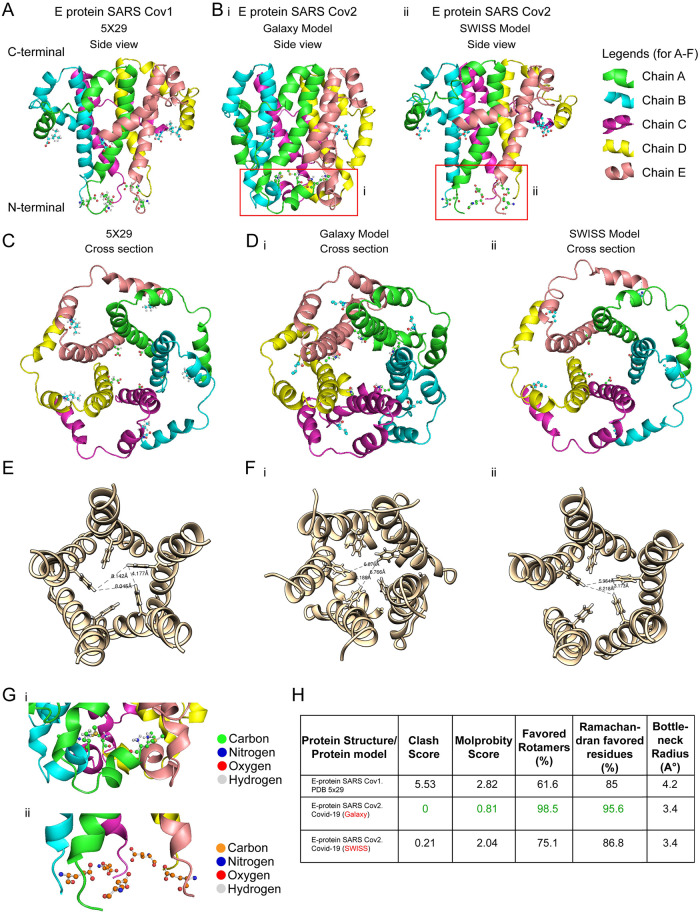
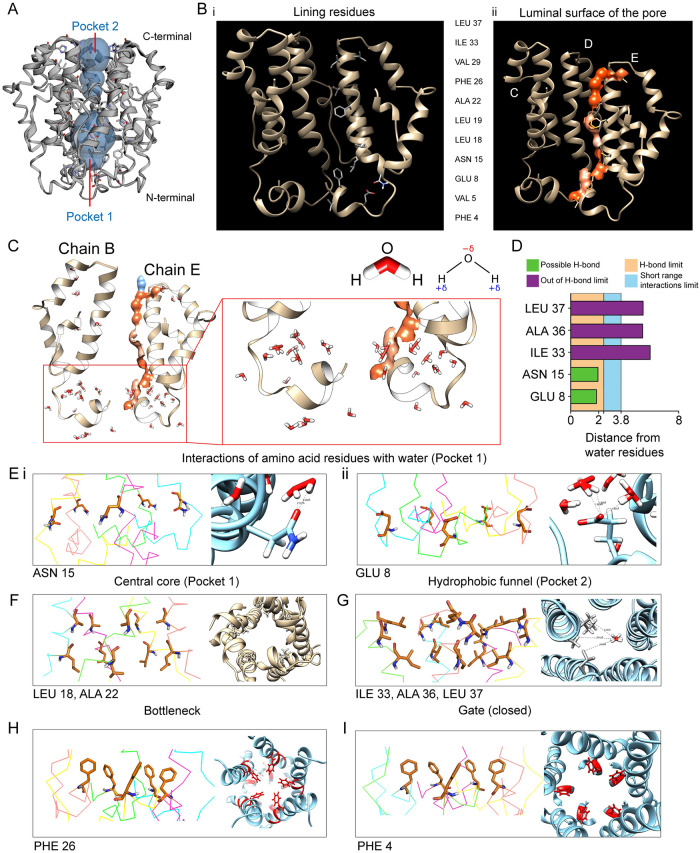
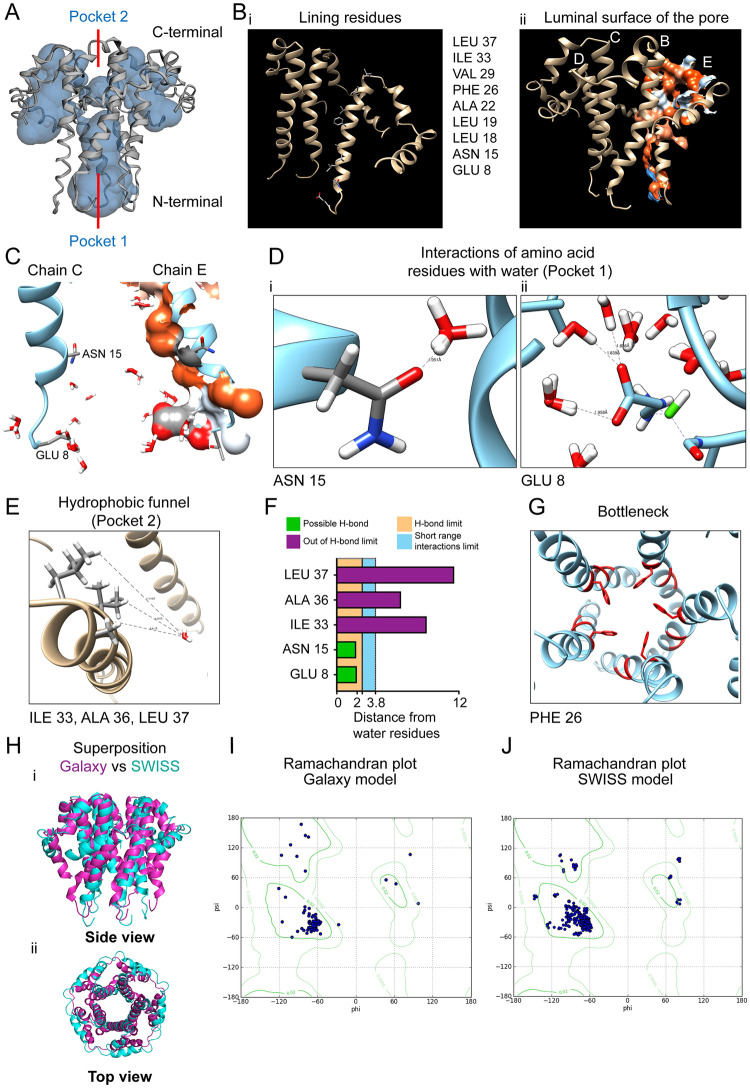
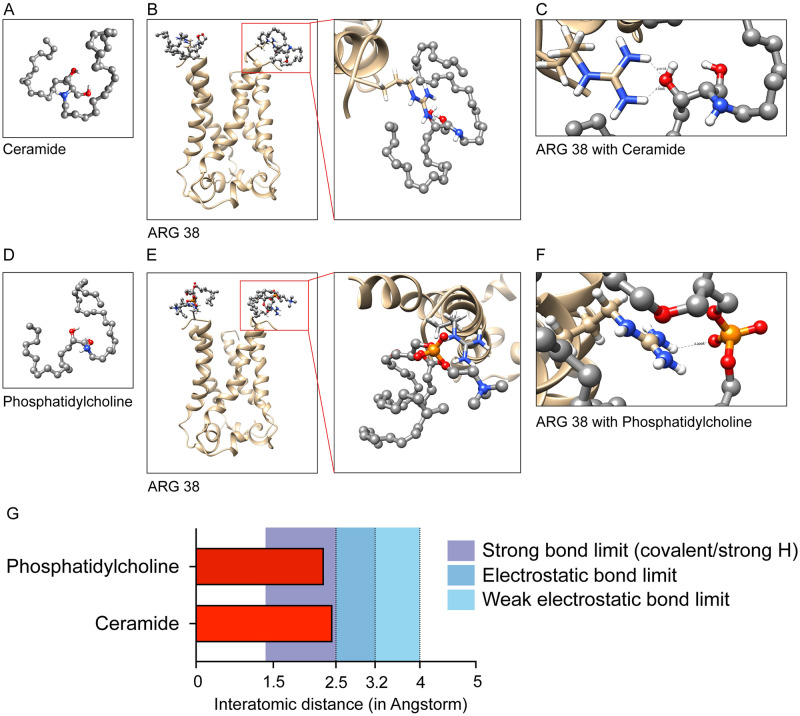
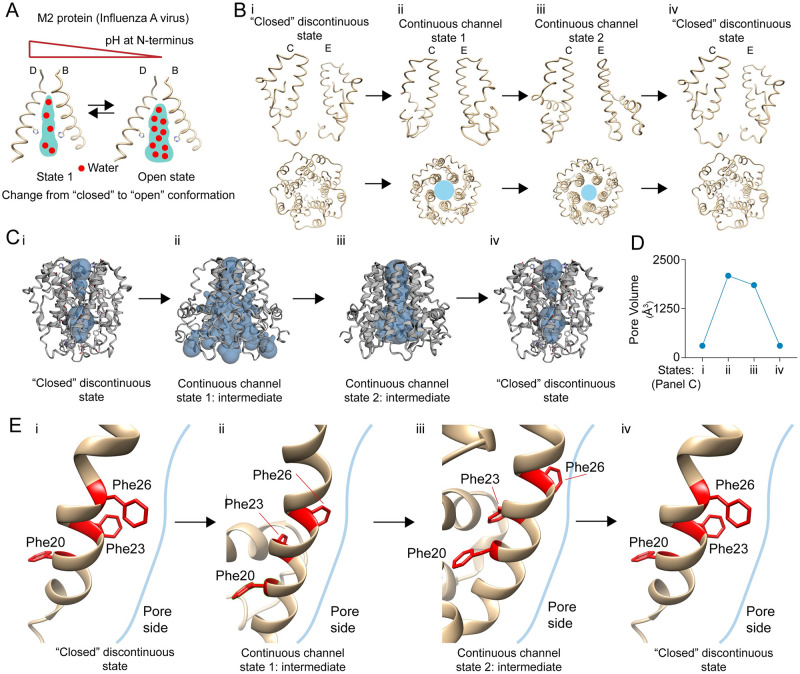
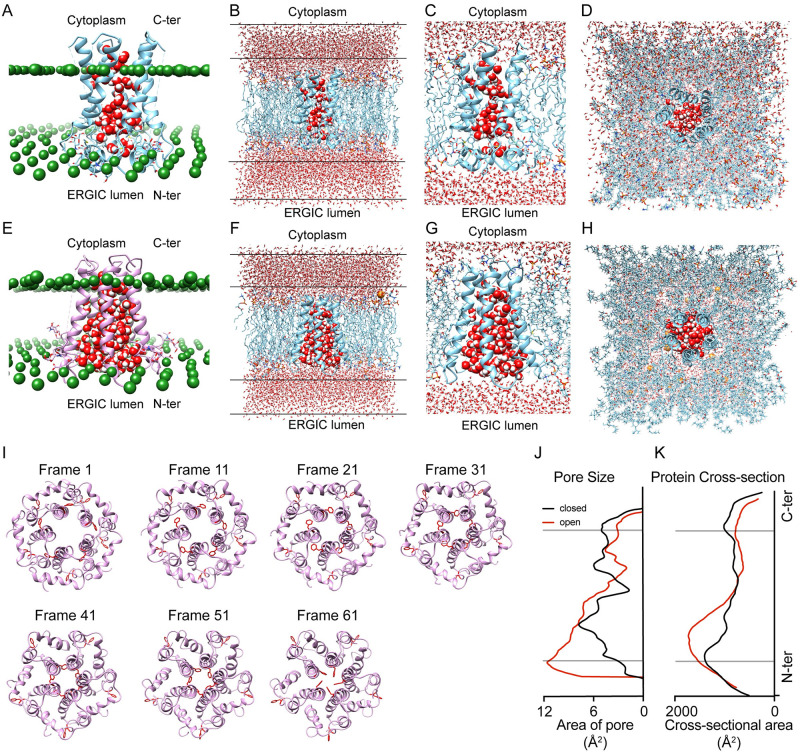
The outbreak of COVID-19 across the world has posed unprecedented and global challenges on multiple fronts. Most of the vaccine and drug development has focused on the spike proteins and viral RNA-polymerases and main protease for viral replication. Using the bioinformatics and structural modelling approach, we modelled the structure of the envelope (E)-protein of novel SARS-CoV-2. The E-protein of this virus shares sequence similarity with that of SARS- CoV-1, and is highly conserved in the N-terminus regions. Incidentally, compared to spike proteins, E proteins demonstrate lower disparity and mutability among the isolated sequences. Using homology modelling, we found that the most favorable structure could function as a gated ion channel conducting H+ ions. Combining pocket estimation and docking with water, we determined that GLU 8 and ASN 15 in the N-terminal region were in close proximity to form H-bonds which was further validated by insertion of the E protein in an ERGIC-mimic membrane. Additionally, two distinct "core" structures were visible, the hydrophobic core and the central core, which may regulate the opening/closing of the channel. We propose this as a mechanism of viral ion channeling activity which plays a critical role in viral infection and pathogenesis. In addition, it provides a structural basis and additional avenues for vaccine development and generating therapeutic interventions against the virus.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- W.H.O. T. Coronavirus disease (COVID-2019) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situatio...
-
- Worldometer. Coronavirus Pandemic 2019 Worldometer. https://www.worldometers.info/coronavirus/
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
