

Identification of MALT1 feedback mechanisms enables rational design of potent antilymphoma regimens for ABC-DLBCL
- PMID: 32785655
- PMCID: PMC7885826
- DOI: 10.1182/blood.2019004713
Identification of MALT1 feedback mechanisms enables rational design of potent antilymphoma regimens for ABC-DLBCL
Abstract
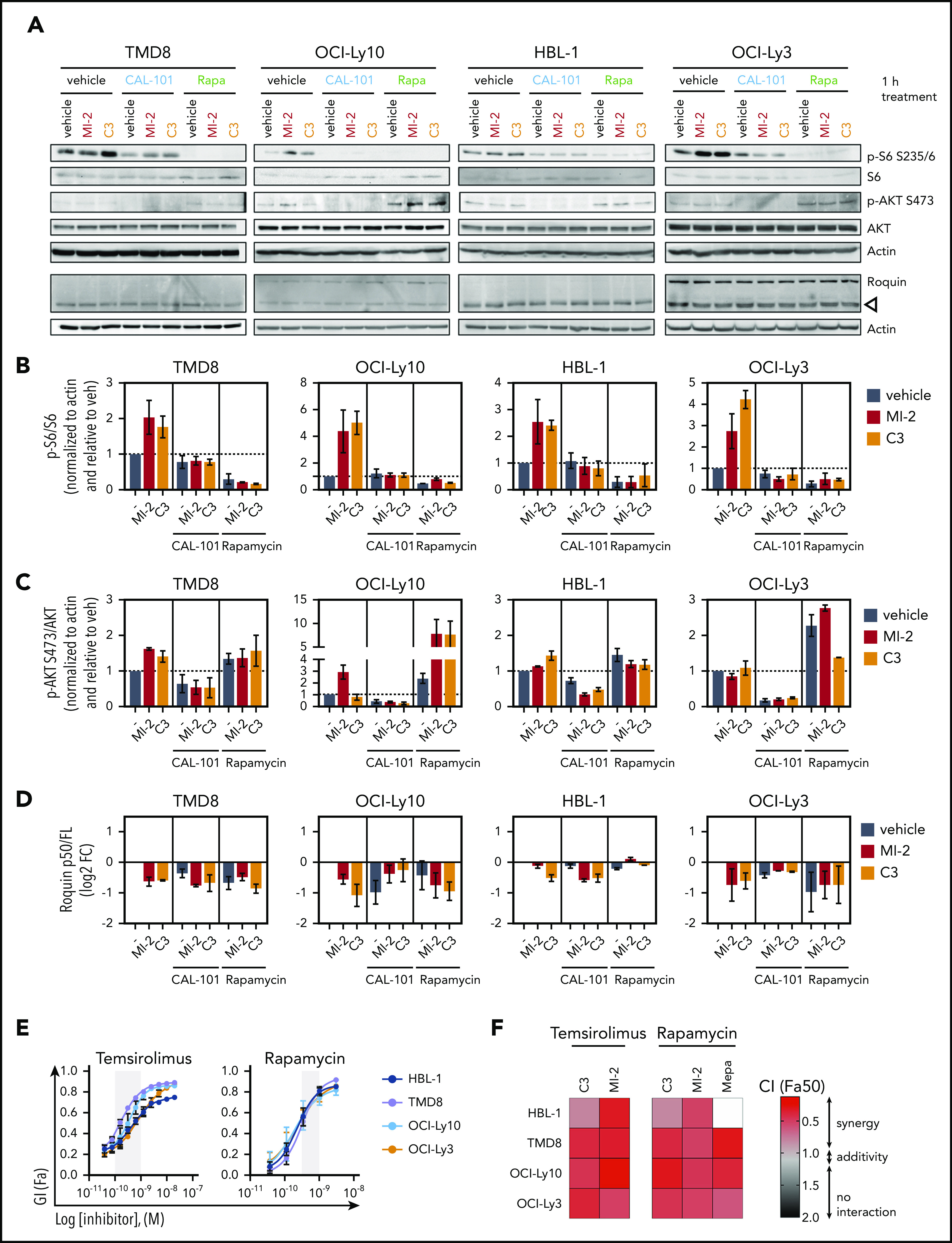
MALT1 inhibitors are promising therapeutic agents for B-cell lymphomas that are dependent on constitutive or aberrant signaling pathways. However, a potential limitation for signal transduction-targeted therapies is the occurrence of feedback mechanisms that enable escape from the full impact of such drugs. Here, we used a functional genomics screen in activated B-cell-like (ABC) diffuse large B-cell lymphoma (DLBCL) cells treated with a small molecule irreversible inhibitor of MALT1 to identify genes that might confer resistance or enhance the activity of MALT1 inhibition (MALT1i). We find that loss of B-cell receptor (BCR)- and phosphatidylinositol 3-kinase (PI3K)-activating proteins enhanced sensitivity, whereas loss of negative regulators of these pathways (eg, TRAF2, TNFAIP3) promoted resistance. These findings were validated by knockdown of individual genes and a combinatorial drug screen focused on BCR and PI3K pathway-targeting drugs. Among these, the most potent combinatorial effect was observed with PI3Kδ inhibitors against ABC-DLBCLs in vitro and in vivo, but that led to an adaptive increase in phosphorylated S6 and eventual disease progression. Along these lines, MALT1i promoted increased MTORC1 activity and phosphorylation of S6K1-T389 and S6-S235/6, an effect that was only partially blocked by PI3Kδ inhibition in vitro and in vivo. In contrast, simultaneous inhibition of MALT1 and MTORC1 prevented S6 phosphorylation, yielded potent activity against DLBCL cell lines and primary patient specimens, and resulted in more profound tumor regression and significantly improved survival of ABC-DLBCLs in vivo compared with PI3K inhibitors. These findings provide a basis for maximal therapeutic impact of MALT1 inhibitors in the clinic, by disrupting feedback mechanisms that might otherwise limit their efficacy.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosures: A.M. receives research support from Janssen Pharmaceuticals and is a consultant for Epizyme and Constellation. The remaining authors declare no competing financial interests.
Figures








Comment in
-
Synergy of MALT1 and mTOR inhibition in DLBCL.Blood. 2021 Feb 11;137(6):724-725. doi: 10.1182/blood.2020008465. Blood. 2021. PMID: 33570607 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
