

Causal Genetic Variants in Stillbirth
- PMID: 32786180
- PMCID: PMC7604888
- DOI: 10.1056/NEJMoa1908753
Causal Genetic Variants in Stillbirth
Abstract
Background: In the majority of cases, the cause of stillbirth remains unknown despite detailed clinical and laboratory evaluation. Approximately 10 to 20% of stillbirths are attributed to chromosomal abnormalities. However, the causal nature of single-nucleotide variants and small insertions and deletions in exomes has been understudied.
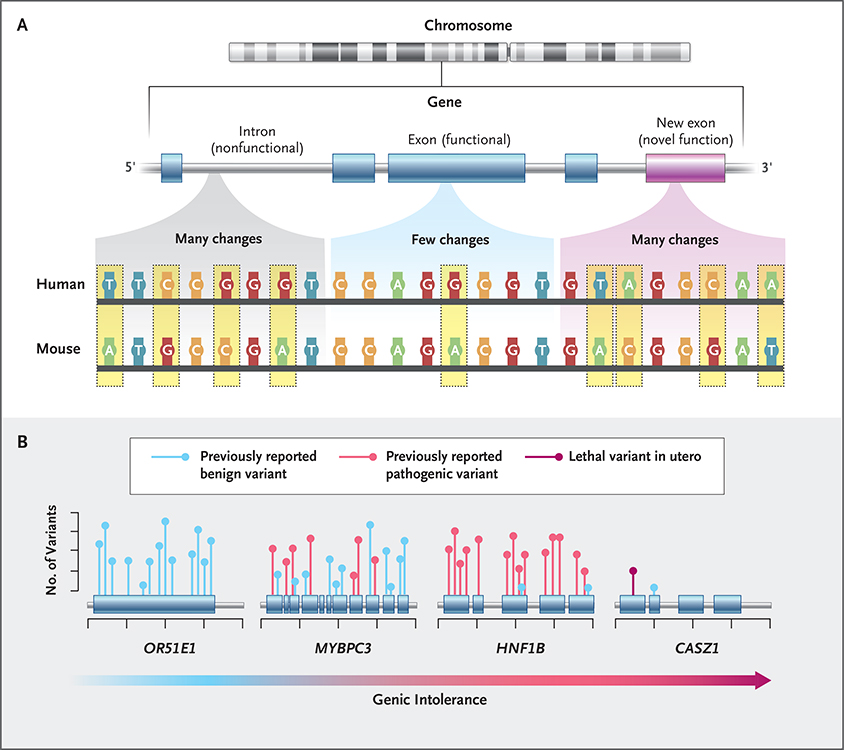
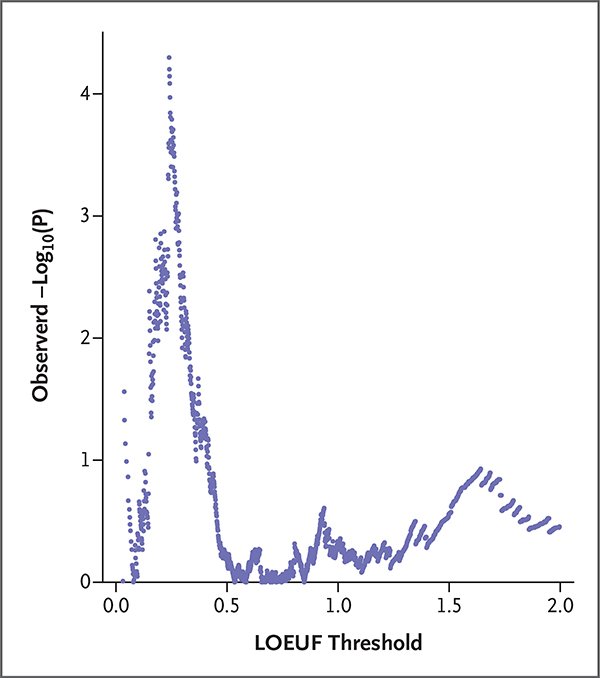
Methods: We generated exome sequencing data for 246 stillborn cases and followed established guidelines to identify causal variants in disease-associated genes. These genes included those that have been associated with stillbirth and strong candidate genes. We also evaluated the contribution of 18,653 genes in case-control analyses stratified according to the degree of depletion of functional variation (described here as "intolerance" to variation).
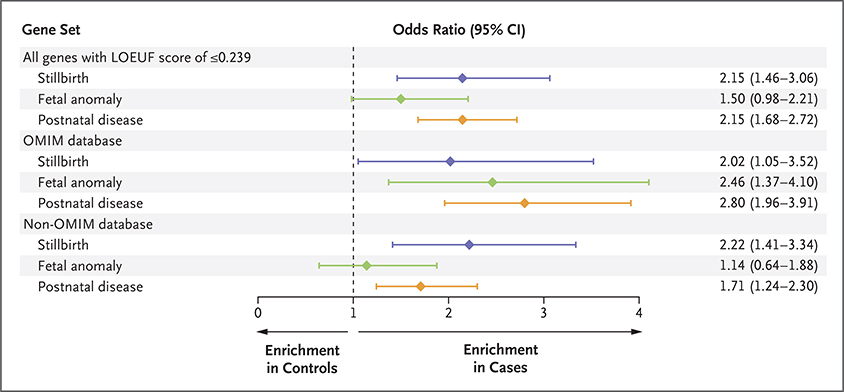
Results: We identified molecular diagnoses in 15 of 246 cases of stillbirth (6.1%) involving seven genes that have been implicated in stillbirth and six disease genes that are good candidates for phenotypic expansion. Among the cases we evaluated, we also found an enrichment of loss-of-function variants in genes that are intolerant to such variation in the human population (odds ratio, 2.15; 95% confidence interval [CI], 1.46 to 3.06). Loss-of-function variants in intolerant genes were concentrated in genes that have not been associated with human disease (odds ratio, 2.22; 95% CI, 1.41 to 3.34), findings that differ from those in two postnatal clinical populations that were also evaluated in this study.
Conclusions: Our findings establish the diagnostic utility of clinical exome sequencing to evaluate the role of small genomic changes in stillbirth. The strength of the novel risk signal (as generated through the stratified analysis) was similar to that in known disease genes, which indicates that the genetic cause of stillbirth remains largely unknown. (Funded by the Institute for Genomic Medicine.).
Copyright © 2020 Massachusetts Medical Society.
Figures
Comment in
-
Genomic Insights into Stillbirth.N Engl J Med. 2020 Sep 17;383(12):1182-1183. doi: 10.1056/NEJMe2016410. Epub 2020 Aug 12. N Engl J Med. 2020. PMID: 32786182 No abstract available.
-
Causal Genetic Variants in Stillbirth.N Engl J Med. 2020 Dec 31;383(27):2687. doi: 10.1056/NEJMc2032136. N Engl J Med. 2020. PMID: 33382937 No abstract available.
References
-
- Wapner RJ, Lewis D. Genetics and metabolic causes of stillbirth. Semin Perinatol 2002;26:70–4. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical