

Robust Sequence Determinants of α-Synuclein Toxicity in Yeast Implicate Membrane Binding
- PMID: 32786289
- PMCID: PMC7442712
- DOI: 10.1021/acschembio.0c00339
Robust Sequence Determinants of α-Synuclein Toxicity in Yeast Implicate Membrane Binding
Abstract
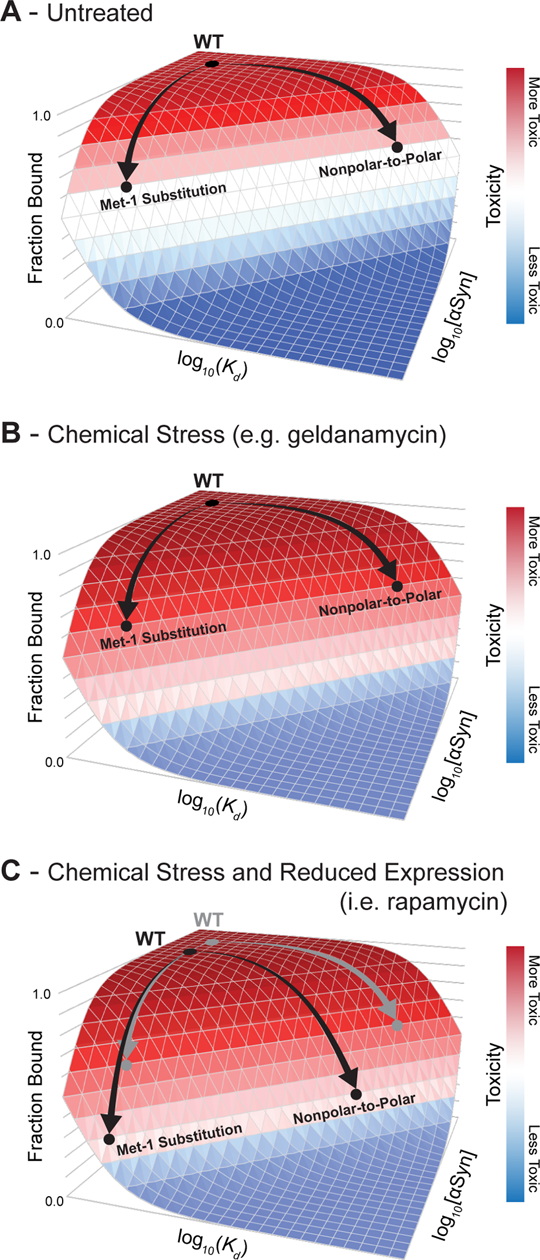
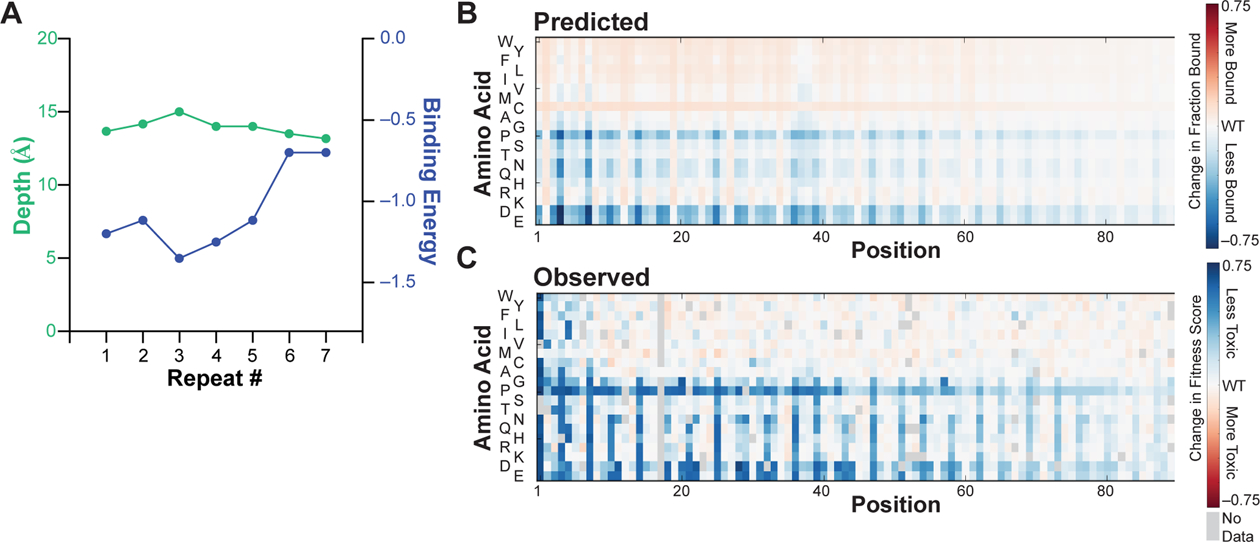
Protein conformations are shaped by cellular environments, but how environmental changes alter the conformational landscapes of specific proteins in vivo remains largely uncharacterized, in part due to the challenge of probing protein structures in living cells. Here, we use deep mutational scanning to investigate how a toxic conformation of α-synuclein, a dynamic protein linked to Parkinson's disease, responds to perturbations of cellular proteostasis. In the context of a course for graduate students in the UCSF Integrative Program in Quantitative Biology, we screened a comprehensive library of α-synuclein missense mutants in yeast cells treated with a variety of small molecules that perturb cellular processes linked to α-synuclein biology and pathobiology. We found that the conformation of α-synuclein previously shown to drive yeast toxicity-an extended, membrane-bound helix-is largely unaffected by these chemical perturbations, underscoring the importance of this conformational state as a driver of cellular toxicity. On the other hand, the chemical perturbations have a significant effect on the ability of mutations to suppress α-synuclein toxicity. Moreover, we find that sequence determinants of α-synuclein toxicity are well described by a simple structural model of the membrane-bound helix. This model predicts that α-synuclein penetrates the membrane to constant depth across its length but that membrane affinity decreases toward the C terminus, which is consistent with orthogonal biophysical measurements. Finally, we discuss how parallelized chemical genetics experiments can provide a robust framework for inquiry-based graduate coursework.
Figures





References
-
- Adkar BV, Tripathi A, Sahoo A, Bajaj K, Goswami D, Chakrabarti P, Swarnkar MK, Gokhale RS, and Varadarajan R (2012) Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure 20, 371–381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 AG002132/AG/NIA NIH HHS/United States
- T32 EB009383/EB/NIBIB NIH HHS/United States
- F31 CA243439/CA/NCI NIH HHS/United States
- U54 CA209891/CA/NCI NIH HHS/United States
- R25 GM056847/GM/NIGMS NIH HHS/United States
- R01 GM117593/GM/NIGMS NIH HHS/United States
- F31 HG011007/HG/NHGRI NIH HHS/United States
- T32 GM008284/GM/NIGMS NIH HHS/United States
- T32 GM067547/GM/NIGMS NIH HHS/United States
- DP2 GM119139/GM/NIGMS NIH HHS/United States
- T32 HL007731/HL/NHLBI NIH HHS/United States
- R35 GM122603/GM/NIGMS NIH HHS/United States
- F31 CA232325/CA/NCI NIH HHS/United States
- T32 GM064337/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
