

Efficient elimination of primary B-ALL cells in vitro and in vivo using a novel 4-1BB-based CAR targeting a membrane-distal CD22 epitope
- PMID: 32788237
- PMCID: PMC7422657
- DOI: 10.1136/jitc-2020-000896
Efficient elimination of primary B-ALL cells in vitro and in vivo using a novel 4-1BB-based CAR targeting a membrane-distal CD22 epitope
Abstract
Background: There are few therapeutic options available for patients with B-cell acute lymphoblastic leukemia (B-ALL) relapsing as CD19- either after chemotherapy or CD19-targeted immunotherapies. CD22-chimeric antigen receptor (CAR) T cells represent an attractive addition to CD19-CAR T cell therapy because they will target both CD22+CD19- B-ALL relapses and CD19- preleukemic cells. However, the immune escape mechanisms from CD22-CAR T cells, and the potential contribution of the epitope binding of the anti-CD22 single-chain variable fragment (scFv) remain understudied.
Methods: Here, we have developed and comprehensively characterized a novel CD22-CAR (clone hCD22.7) targeting a membrane-distal CD22 epitope and tested its cytotoxic effects against B-ALL cells both in in vitro and in vivo assays.
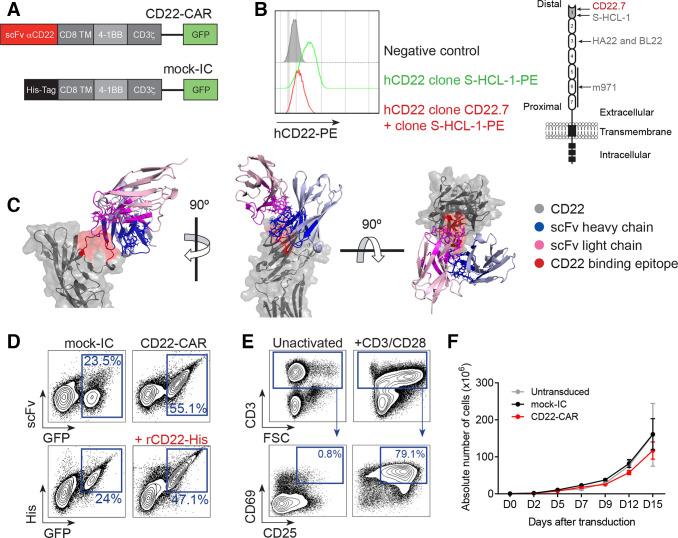
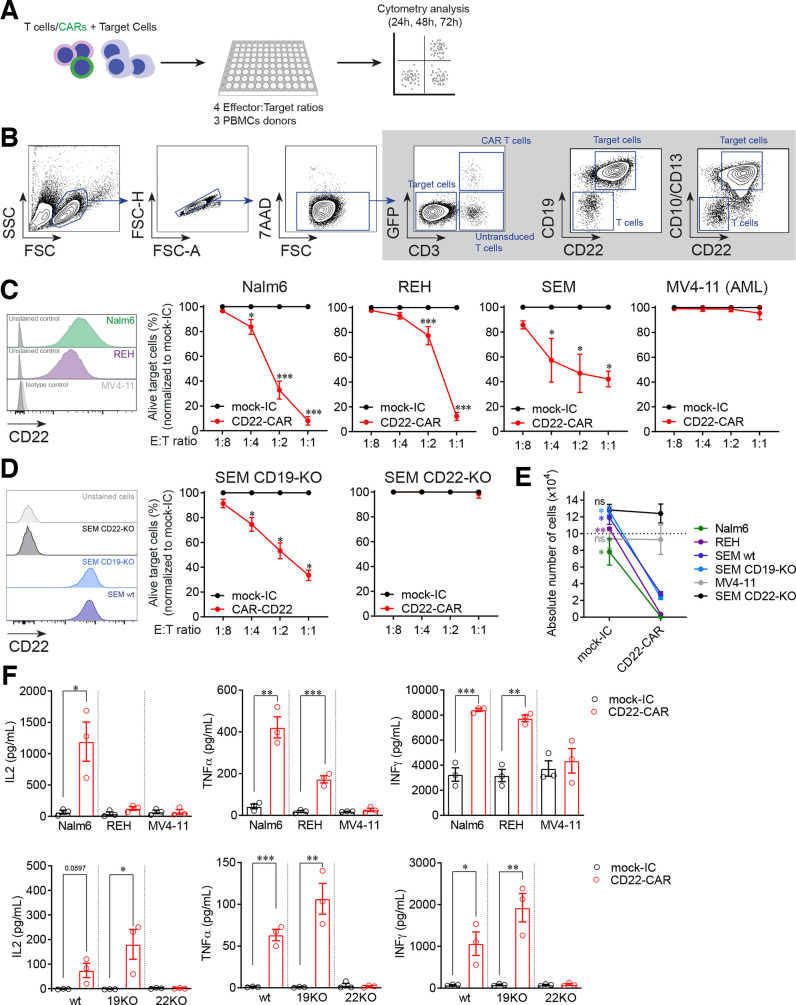
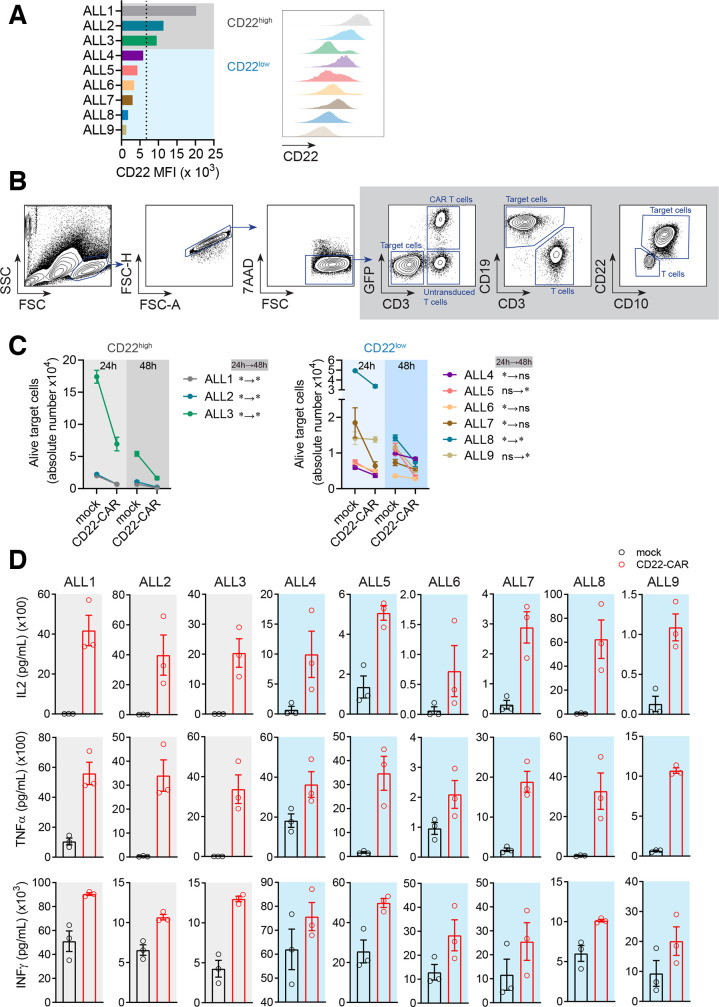
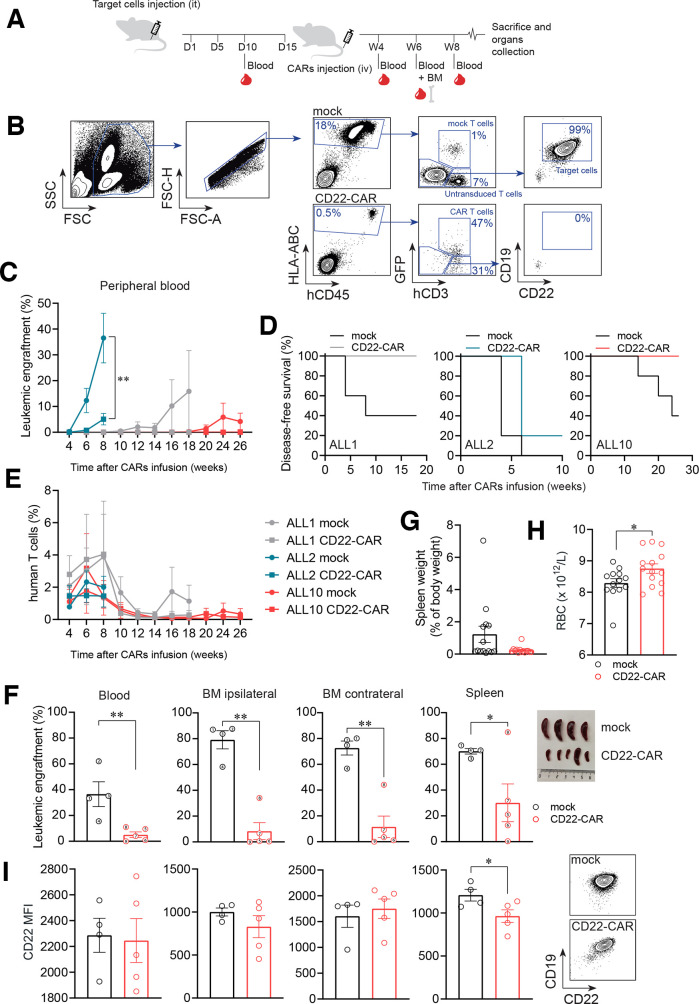
Results: Conformational epitope mapping, cross-blocking, and molecular docking assays revealed that the hCD22.7 scFv is a high-affinity binding antibody which specifically binds to the ESTKDGKVP sequence, located in the Ig-like V-type domain, the most distal domain of CD22. We observed efficient killing of B-ALL cells in vitro, although the kinetics were dependent on the level of CD22 expression. Importantly, we show an efficient in vivo control of patients with B-ALL derived xenografts with diverse aggressiveness, coupled to long-term hCD22.7-CAR T cell persistence. Remaining leukemic cells at sacrifice maintained full expression of CD22, ruling out CAR pressure-mediated antigen loss. Finally, the immunogenicity capacity of this hCD22.7-scFv was very similar to that of other CD22 scFv previously used in adoptive T cell therapy.
Conclusions: We report a novel, high-affinity hCD22.7 scFv which targets a membrane-distal epitope of CD22. 4-1BB-based hCD22.7-CAR T cells efficiently eliminate clinically relevant B- CD22high and CD22low ALL primary samples in vitro and in vivo. Our study supports the clinical translation of this hCD22.7-CAR as either single or tandem CD22-CD19-CAR for both naive and anti-CD19-resistant patients with B-ALL.
Keywords: T-lymphocytes; cell engineering; hematologic neoplasms; immunotherapy; receptors, chimeric antigen.
© Author(s) (or their employer(s)) 2020. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: PM is co-founder of OneChain Immunotherapeutics (OCI), a IJC spin-off company focused on the development of CAR T-cell therapies. The patent of the membrane-distal CD22 scFv clone has been licenced to OCI.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials