

Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling
- PMID: 32788668
- PMCID: PMC7878580
- DOI: 10.1038/s41587-020-0645-6
Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling
Erratum in
-
Author Correction: Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling.Nat Biotechnol. 2023 Sep;41(9):1345. doi: 10.1038/s41587-023-01942-1. Nat Biotechnol. 2023. PMID: 37653227 No abstract available.
Abstract
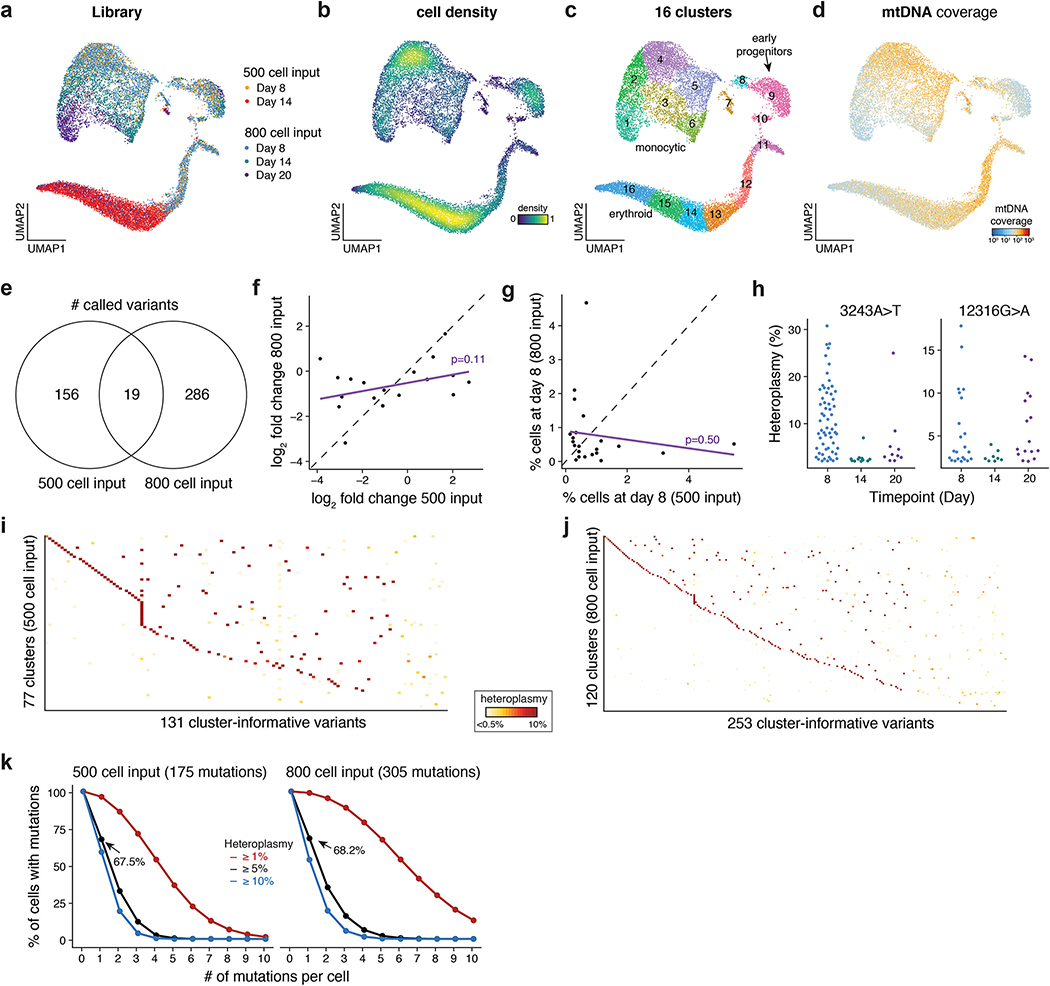
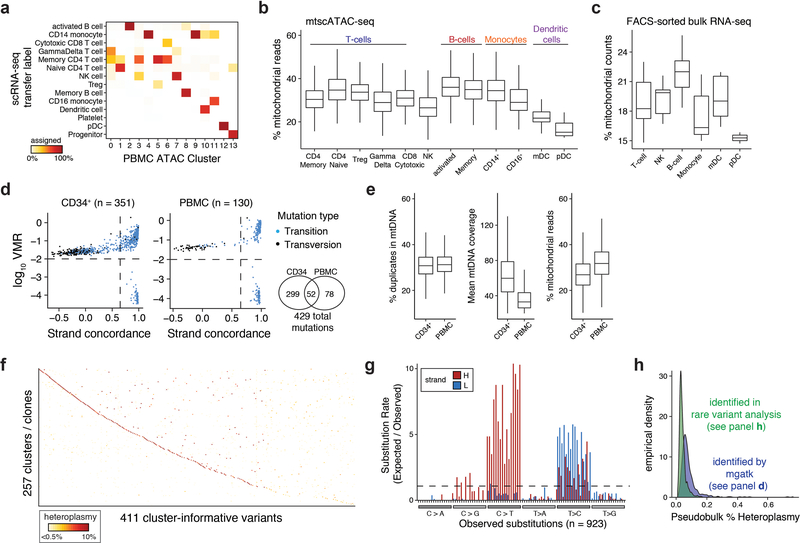
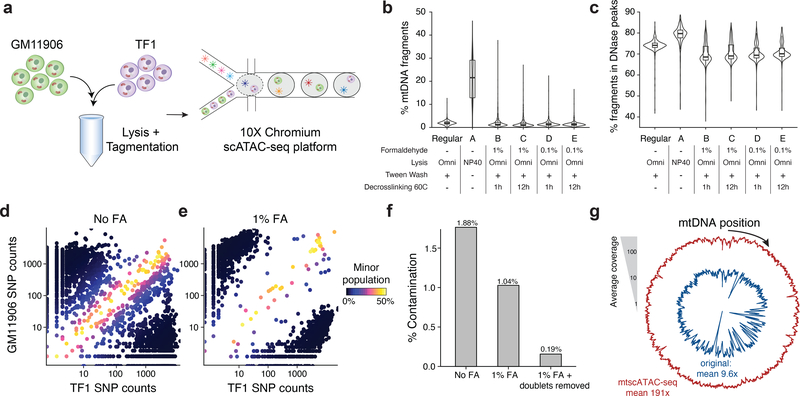
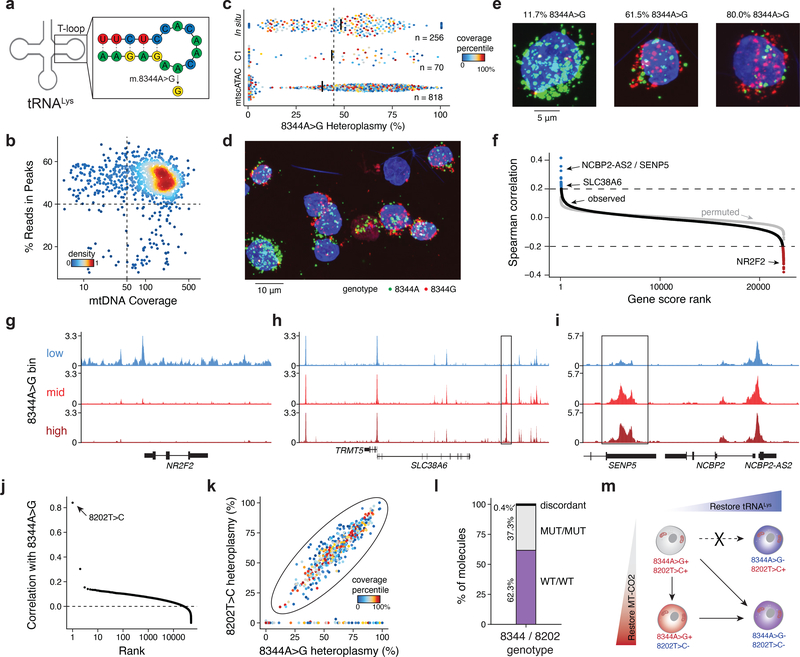
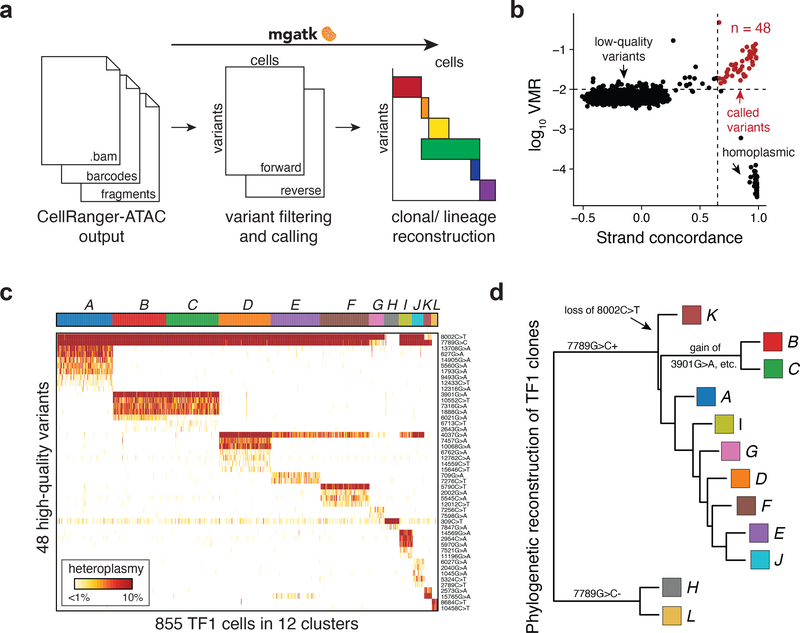
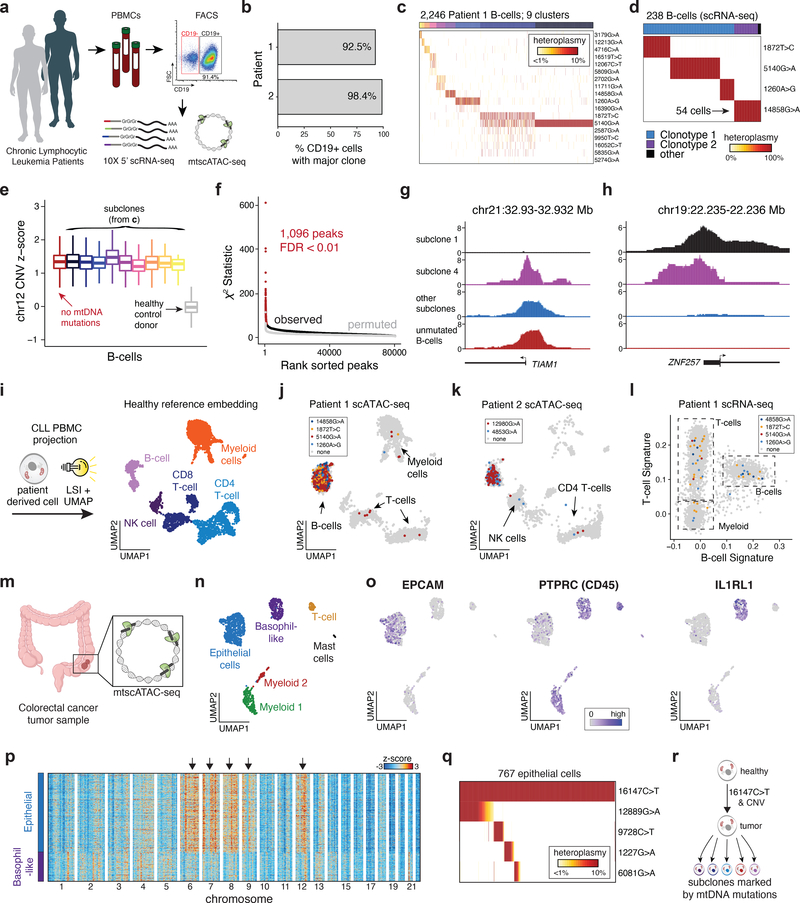
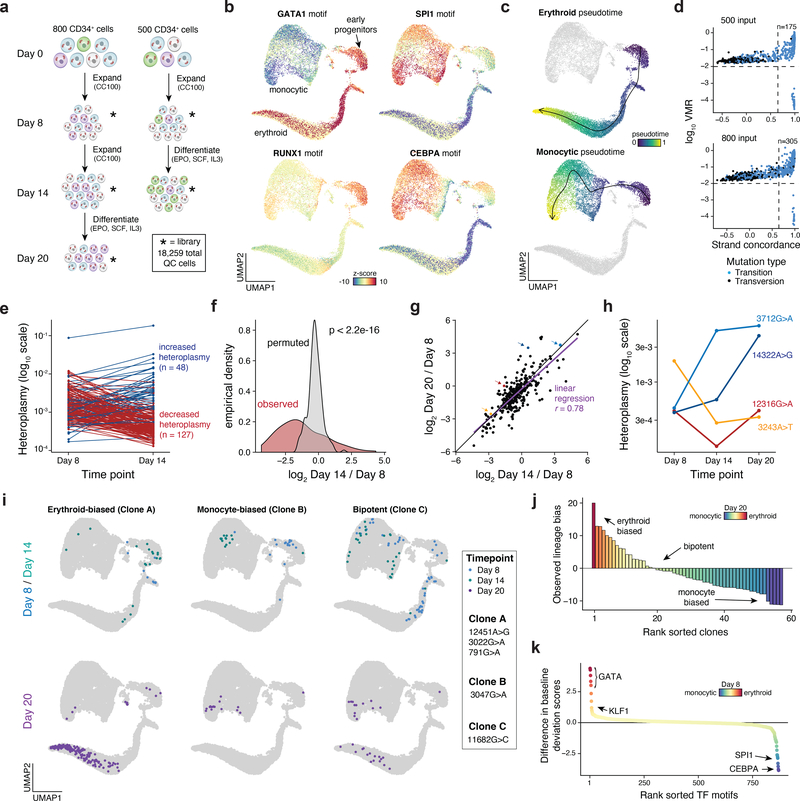
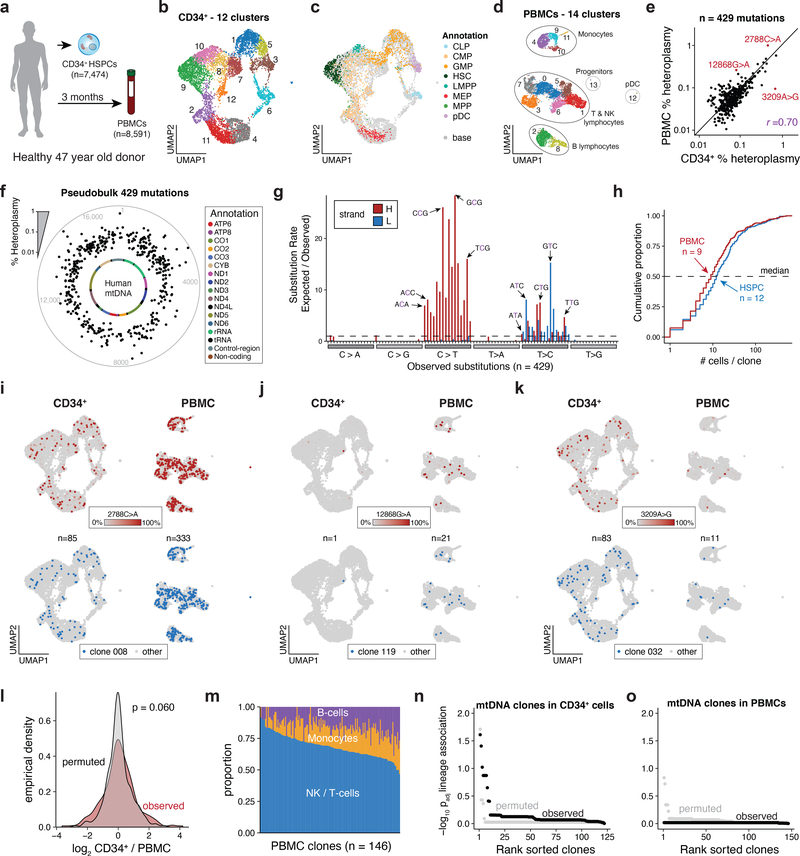
Natural mitochondrial DNA (mtDNA) mutations enable the inference of clonal relationships among cells. mtDNA can be profiled along with measures of cell state, but has not yet been combined with the massively parallel approaches needed to tackle the complexity of human tissue. Here, we introduce a high-throughput, droplet-based mitochondrial single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq), a method that combines high-confidence mtDNA mutation calling in thousands of single cells with their concomitant high-quality accessible chromatin profile. This enables the inference of mtDNA heteroplasmy, clonal relationships, cell state and accessible chromatin variation in individual cells. We reveal single-cell variation in heteroplasmy of a pathologic mtDNA variant, which we associate with intra-individual chromatin variability and clonal evolution. We clonally trace thousands of cells from cancers, linking epigenomic variability to subclonal evolution, and infer cellular dynamics of differentiating hematopoietic cells in vitro and in vivo. Taken together, our approach enables the study of cellular population dynamics and clonal properties in vivo.
Figures
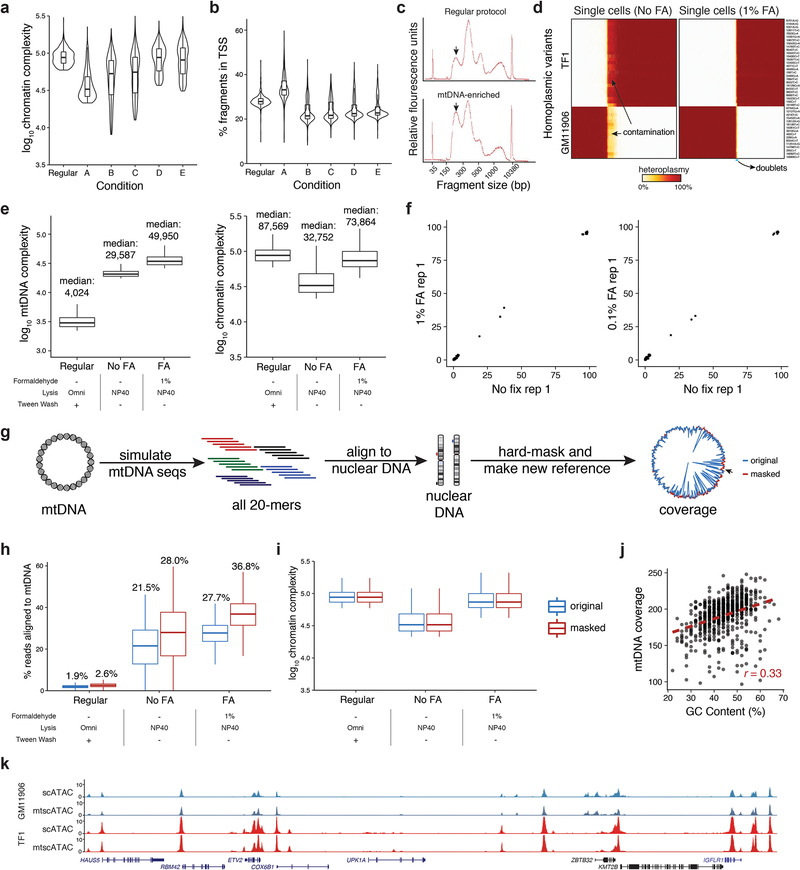
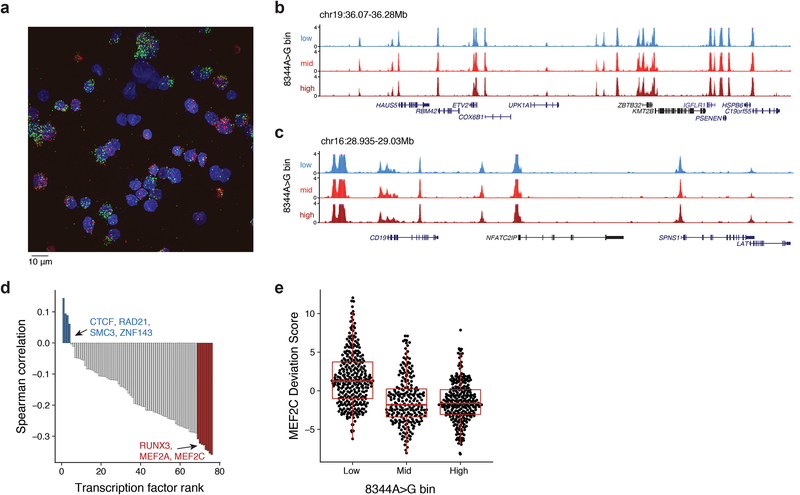
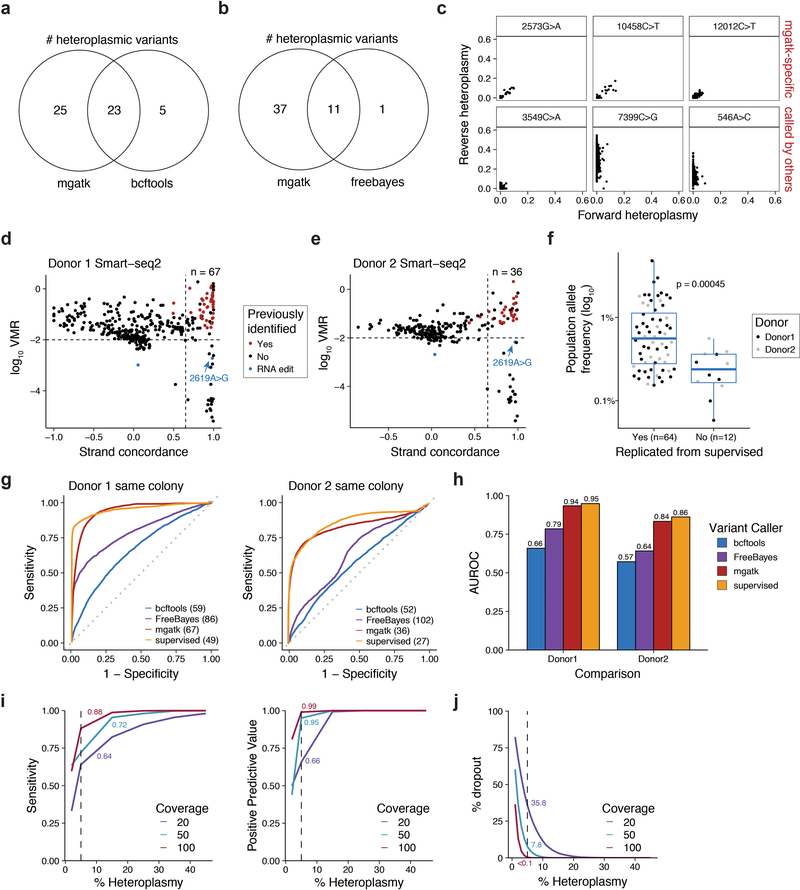
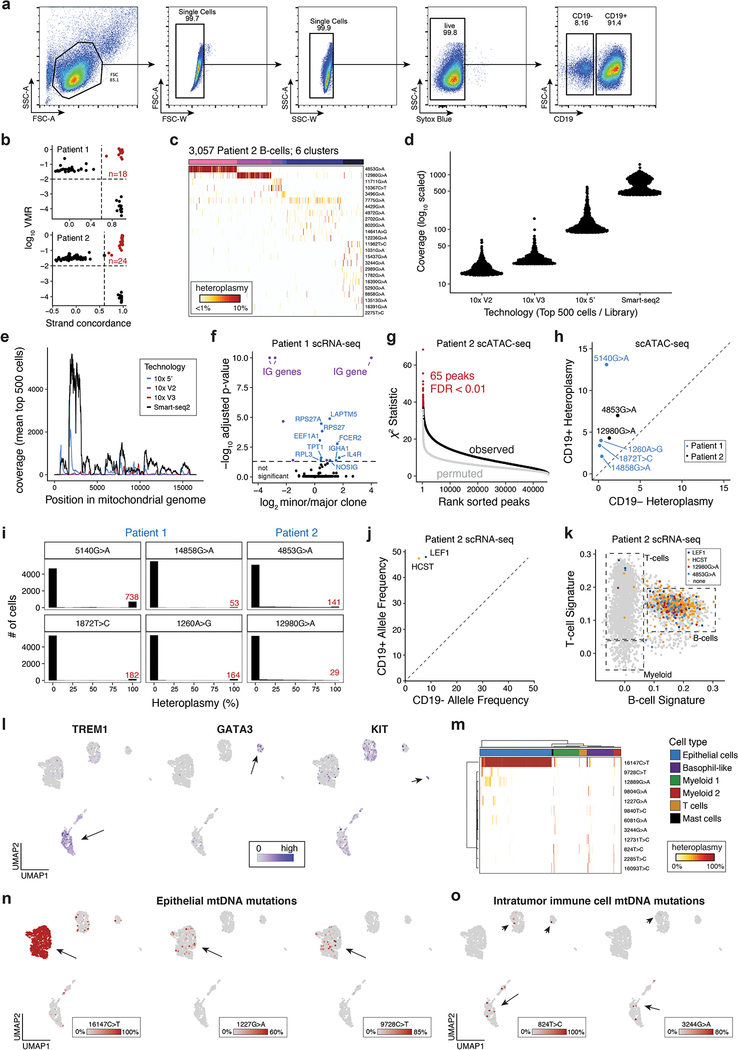
References
-
- Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet 16, 530–542 (2015). - PubMed
-
- Kang E et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell 18, 625–636 (2016). - PubMed
METHODS ONLY REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
