

scRepertoire: An R-based toolkit for single-cell immune receptor analysis
- PMID: 32789006
- PMCID: PMC7400693
- DOI: 10.12688/f1000research.22139.2
scRepertoire: An R-based toolkit for single-cell immune receptor analysis
Abstract
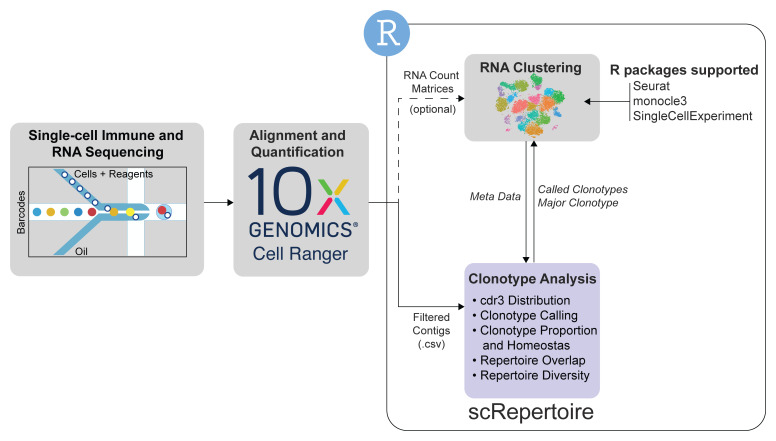
Single-cell sequencing is an emerging technology in the field of immunology and oncology that allows researchers to couple RNA quantification and other modalities, like immune cell receptor profiling at the level of an individual cell. A number of workflows and software packages have been created to process and analyze single-cell transcriptomic data. These packages allow users to take the vast dimensionality of the data generated in single-cell-based experiments and distill the data into novel insights. Unlike the transcriptomic field, there is a lack of options for software that allow for single-cell immune receptor profiling. Enabling users to easily combine mRNA and immune profiling, scRepertoire was built to process data derived from 10x Genomics Chromium Immune Profiling for both T-cell receptor (TCR) and immunoglobulin (Ig) enrichment workflows and subsequently interacts with a number of popular R packages for single-cell expression, such as Seurat. The scRepertoire R package and processed data are open source and available on GitHub and provides in-depth tutorials on the capability of the package.
Keywords: R; Single-cell RNA sequencing; clonotypic analysis; immune receptor profiling.
Copyright: © 2020 Borcherding N et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
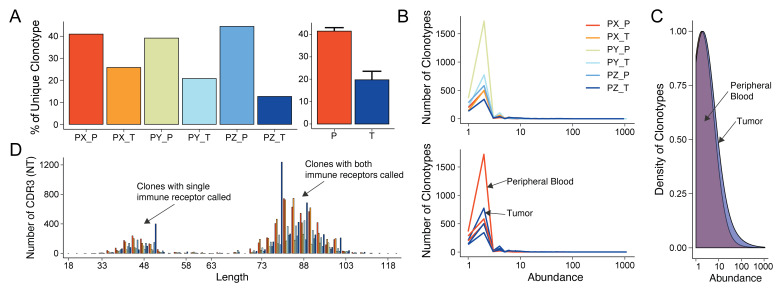
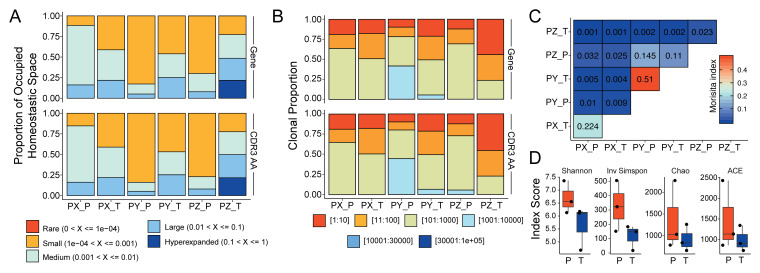
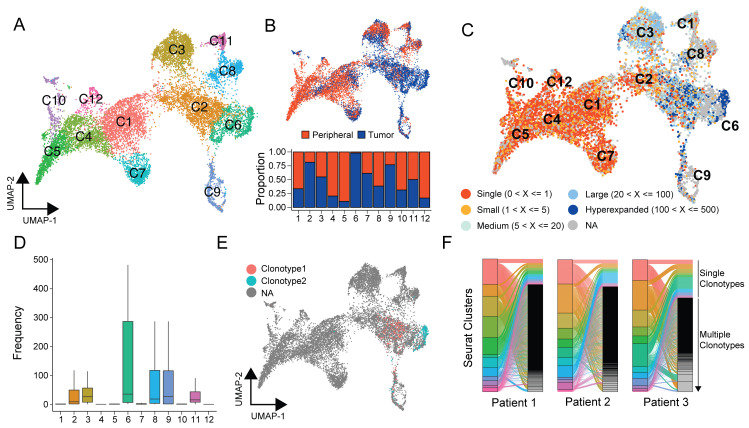
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources