

Genotype networks of 80 quantitative Arabidopsis thaliana phenotypes reveal phenotypic evolvability despite pervasive epistasis
- PMID: 32790763
- PMCID: PMC7447023
- DOI: 10.1371/journal.pcbi.1008082
Genotype networks of 80 quantitative Arabidopsis thaliana phenotypes reveal phenotypic evolvability despite pervasive epistasis
Abstract
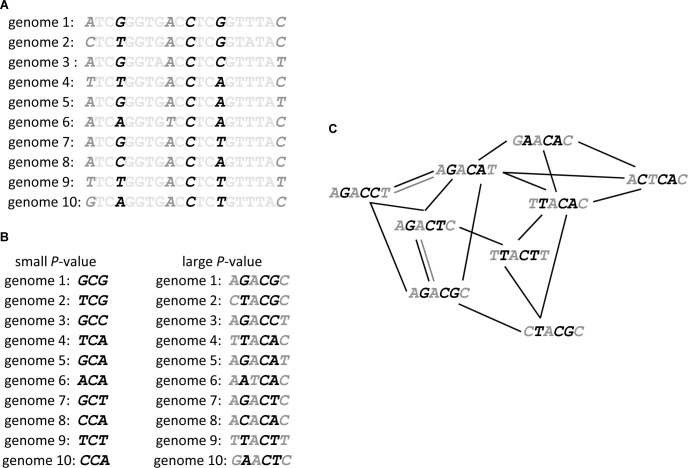
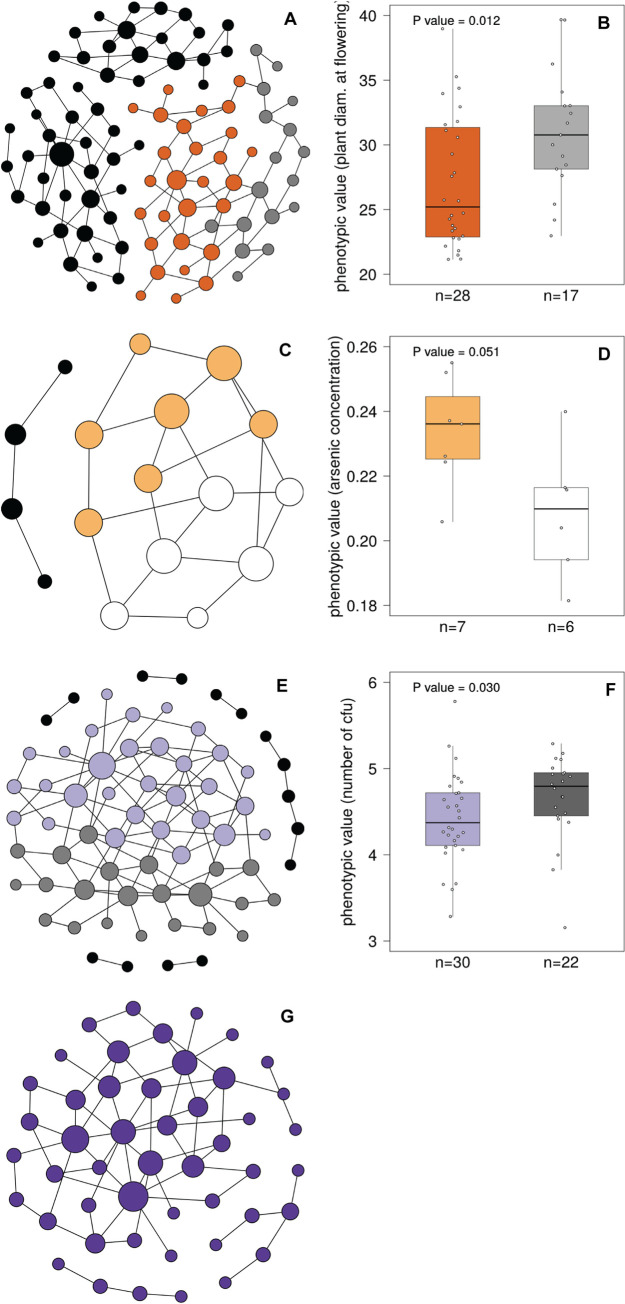
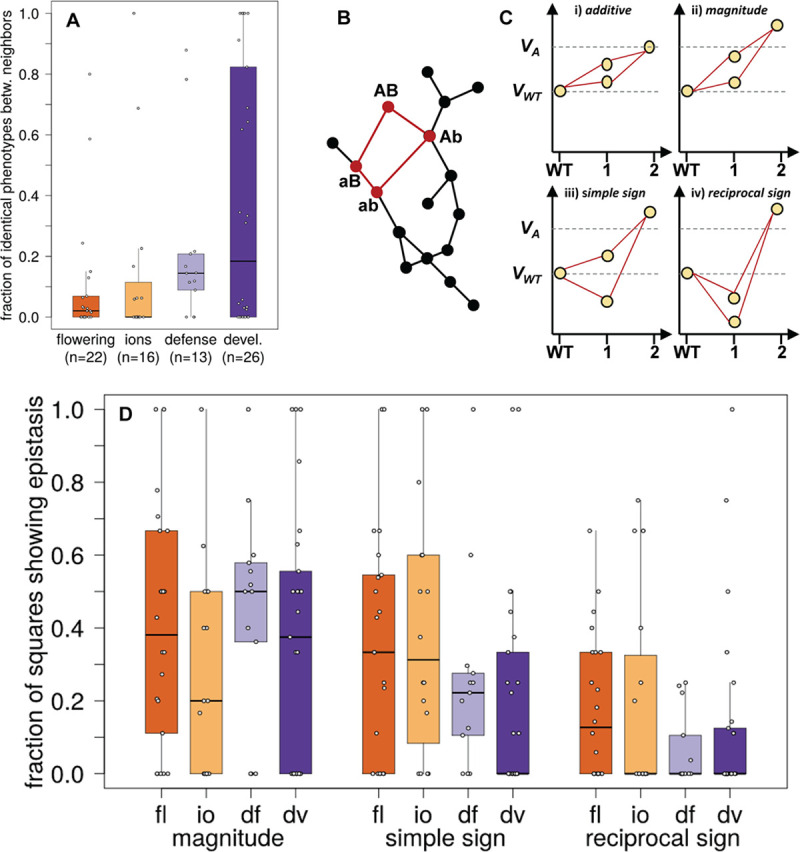
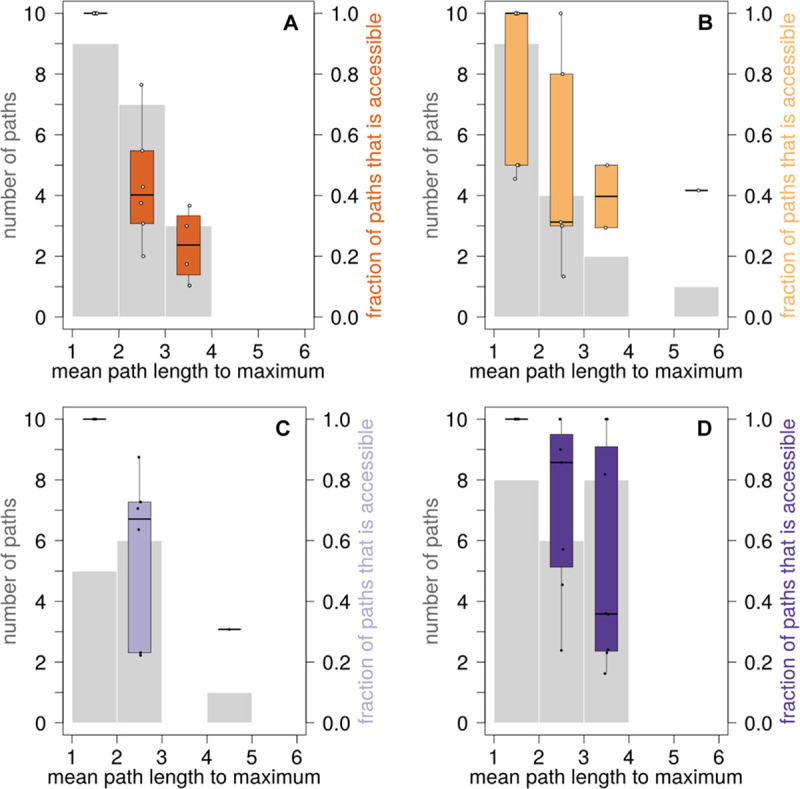
We study the genotype-phenotype maps of 80 quantitative phenotypes in the model plant Arabidopsis thaliana, by representing the genotypes affecting each phenotype as a genotype network. In such a network, each vertex or node corresponds to an individual's genotype at all those genomic loci that affect a given phenotype. Two vertices are connected by an edge if the associated genotypes differ in exactly one nucleotide. The 80 genotype networks we analyze are based on data from genome-wide association studies of 199 A. thaliana accessions. They form connected graphs whose topography differs substantially among phenotypes. We focus our analysis on the incidence of epistasis (non-additive interactions among mutations) because a high incidence of epistasis can reduce the accessibility of evolutionary paths towards high or low phenotypic values. We find epistatic interactions in 67 phenotypes, and in 51 phenotypes every pairwise mutant interaction is epistatic. Moreover, we find phenotype-specific differences in the fraction of accessible mutational paths to maximum phenotypic values. However, even though epistasis affects the accessibility of maximum phenotypic values, the relationships between genotypic and phenotypic change of our analyzed phenotypes are sufficiently smooth that some evolutionary paths remain accessible for most phenotypes, even where epistasis is pervasive. The genotype network representation we use can complement existing approaches to understand the genetic architecture of polygenic traits in many different organisms.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
