

Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia
- PMID: 32793890
- PMCID: PMC7418874
- DOI: 10.1158/0008-5472.BCD-19-0041
Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia
Abstract
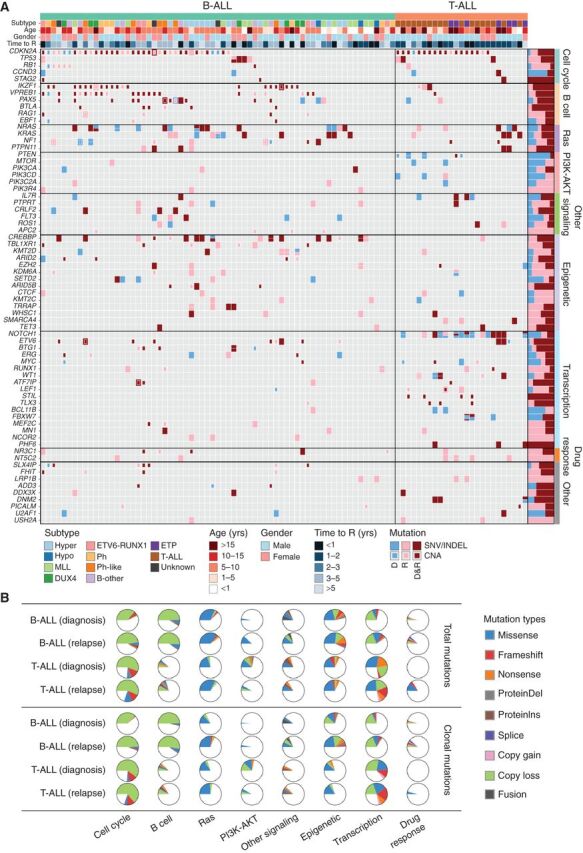
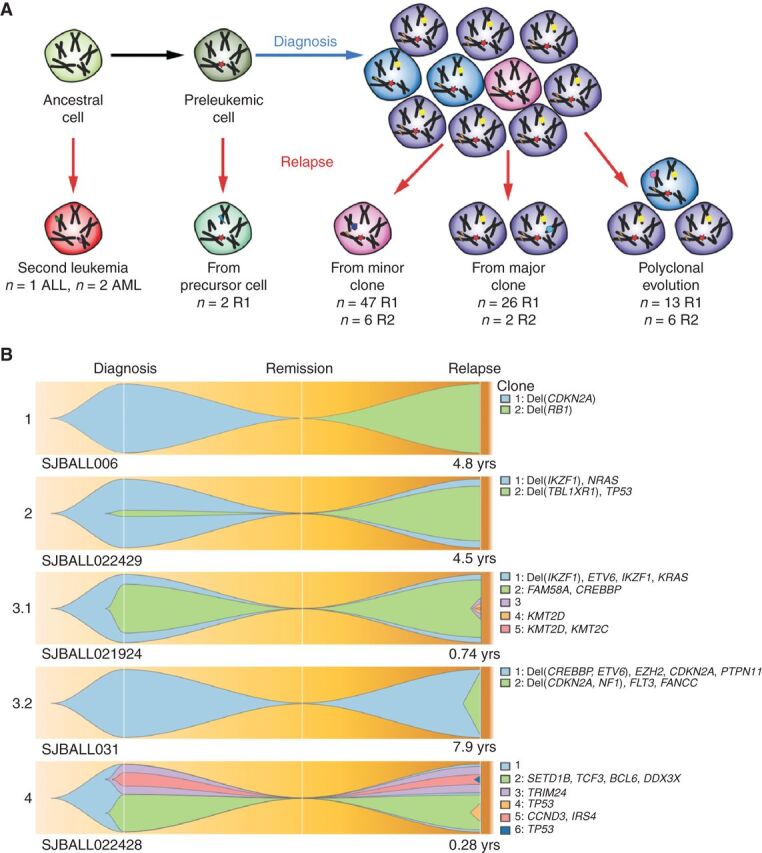
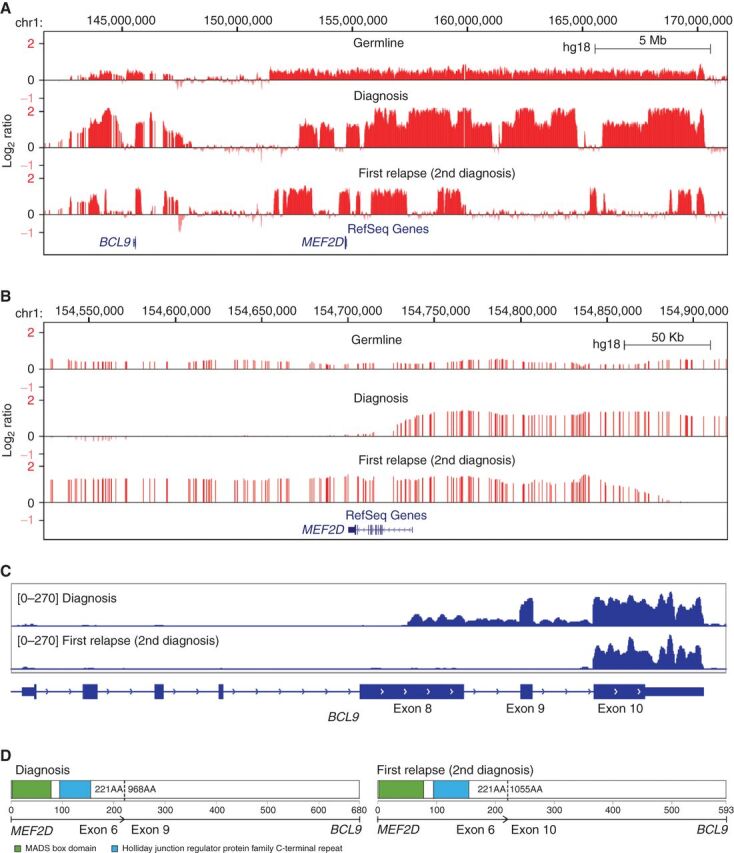
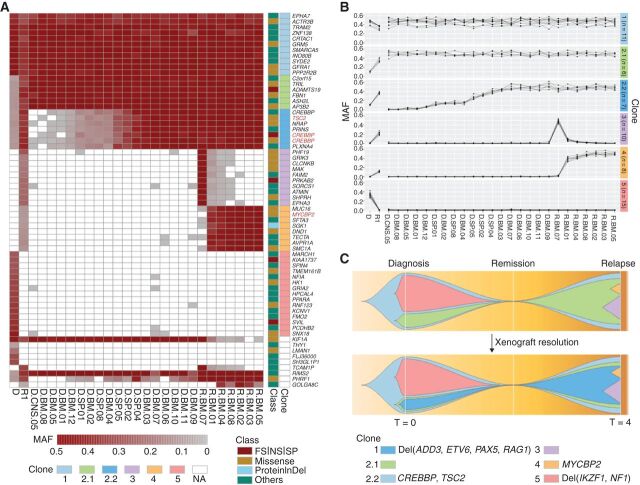
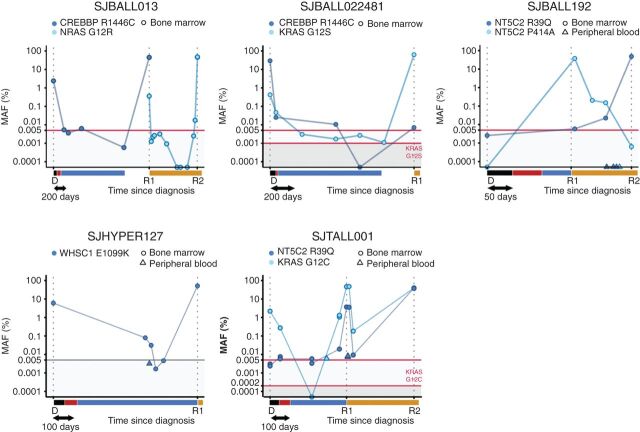
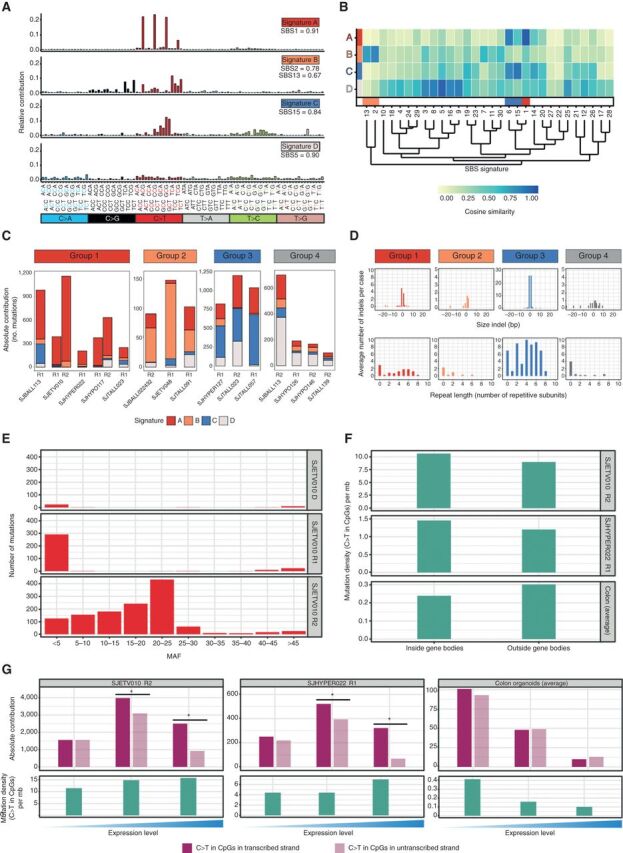
Relapse of acute lymphoblastic leukemia (ALL) remains a leading cause of childhood death. Prior studies have shown clonal mutations at relapse often arise from relapse-fated subclones that exist at diagnosis. However, the genomic landscape, evolutionary trajectories and mutational mechanisms driving relapse are incompletely understood. In an analysis of 92 cases of relapsed childhood ALL, incorporating multimodal DNA and RNA sequencing, deep digital mutational tracking and xenografting to formally define clonal structure, we identify 50 significant targets of mutation with distinct patterns of mutational acquisition or enrichment. CREBBP, NOTCH1, and Ras signaling mutations rose from diagnosis subclones, whereas variants in NCOR2, USH2A and NT5C2 were exclusively observed at relapse. Evolutionary modeling and xenografting demonstrated that relapse-fated clones were minor (50%), major (27%) or multiclonal (18%) at diagnosis. Putative second leukemias, including those with lineage shift, were shown to most commonly represent relapse from an ancestral clone rather than a truly independent second primary leukemia. A subset of leukemias prone to repeated relapse exhibited hypermutation driven by at least three distinct mutational processes, resulting in heightened neoepitope burden and potential vulnerability to immunotherapy. Finally, relapse-driving sequence mutations were detected prior to relapse using deep digital PCR at levels comparable to orthogonal approaches to monitor levels of measurable residual disease. These results provide a genomic framework to anticipate and circumvent relapse by earlier detection and targeting of relapse-fated clones.
Keywords: acute lymphoblastic leukemia; clonal evolution; genomics; hypermutation; neoepitopes; relapse.
Conflict of interest statement
Conflict of interest disclosure: Charles Mullighan: Commercial Research Grant (1. Entity: Abbvie; Relationship: self; Type: Major ($10,000 or more). 2. Entity: Loxo Oncology; Relationship: self; Type: Major ($10,000 or more). 3. Entity: Pfizer, Relationship: self; Type: Major ($10,000 or more)). Honoraria from Speakers Bureau (1. Entity: Amgen; Relationship: self; Type: Minor ($10,000 or less). 2. Pfizer; Relationship: self; Type: Minor ($10,000 or less). Scientific Advisory Board (Entity: Illumina; Relationship: self; Type: Minor ($10,000 or less)); Mary V. Relling, Commercial Research Grant (Entity: Servier Pharmaceuticals; Relationship: self; Type: Major ($10,000 or more)); Paul G. Thomas, Honoraria from Speakers Bureau (1. Entity: Illumina; Relationship: self; Type: Minor ($10,000 or less); 2. Entity: PACT Pharma; Relationship: Myself; Type: Minor ($10,000 or less)).
Figures
Comment in
-
Deciphering the Clonal Origin of Relapsed Acute Lymphoblastic Leukemia in Children.Blood Cancer Discov. 2020 Jun 22;1(1):21-22. doi: 10.1158/2643-3249.BCD-20-0056. eCollection 2020 Jul. Blood Cancer Discov. 2020. PMID: 34661139 Free PMC article.
References
-
- Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med 2015;373:1541–52. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
