

Riemannian geometry and statistical modeling correct for batch effects and control false discoveries in single-cell surface protein count data
- PMID: 32794969
- PMCID: PMC7437020
- DOI: 10.1103/PhysRevE.102.012409
Riemannian geometry and statistical modeling correct for batch effects and control false discoveries in single-cell surface protein count data
Abstract
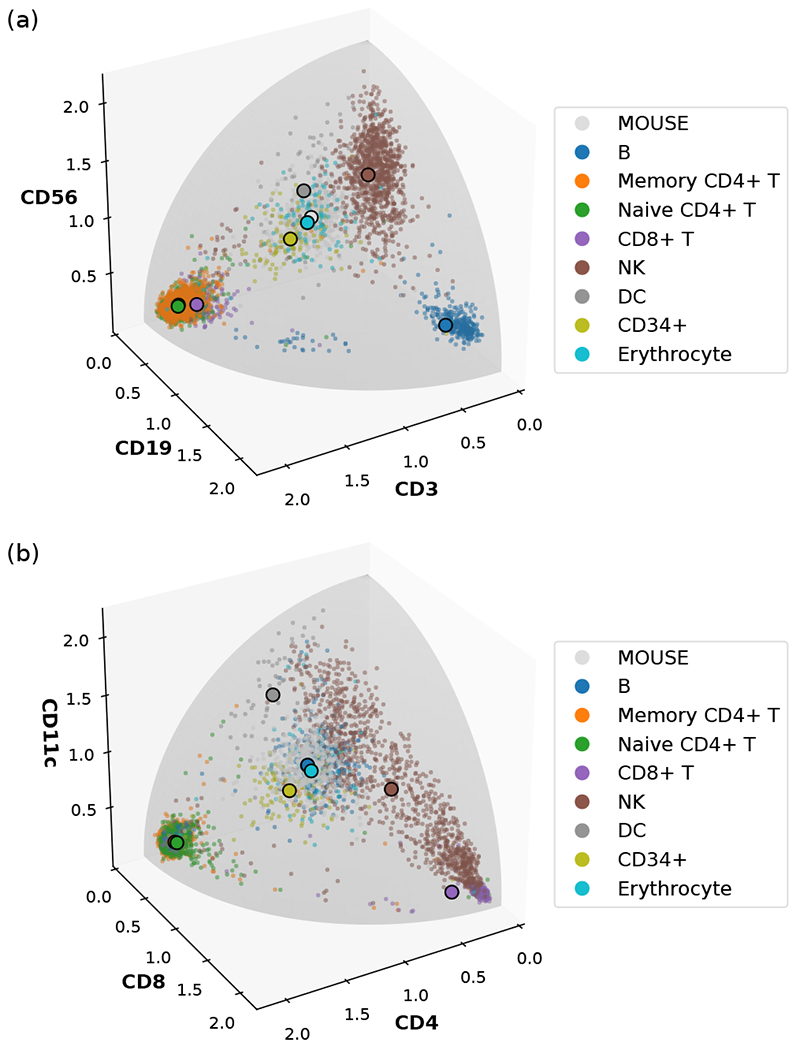
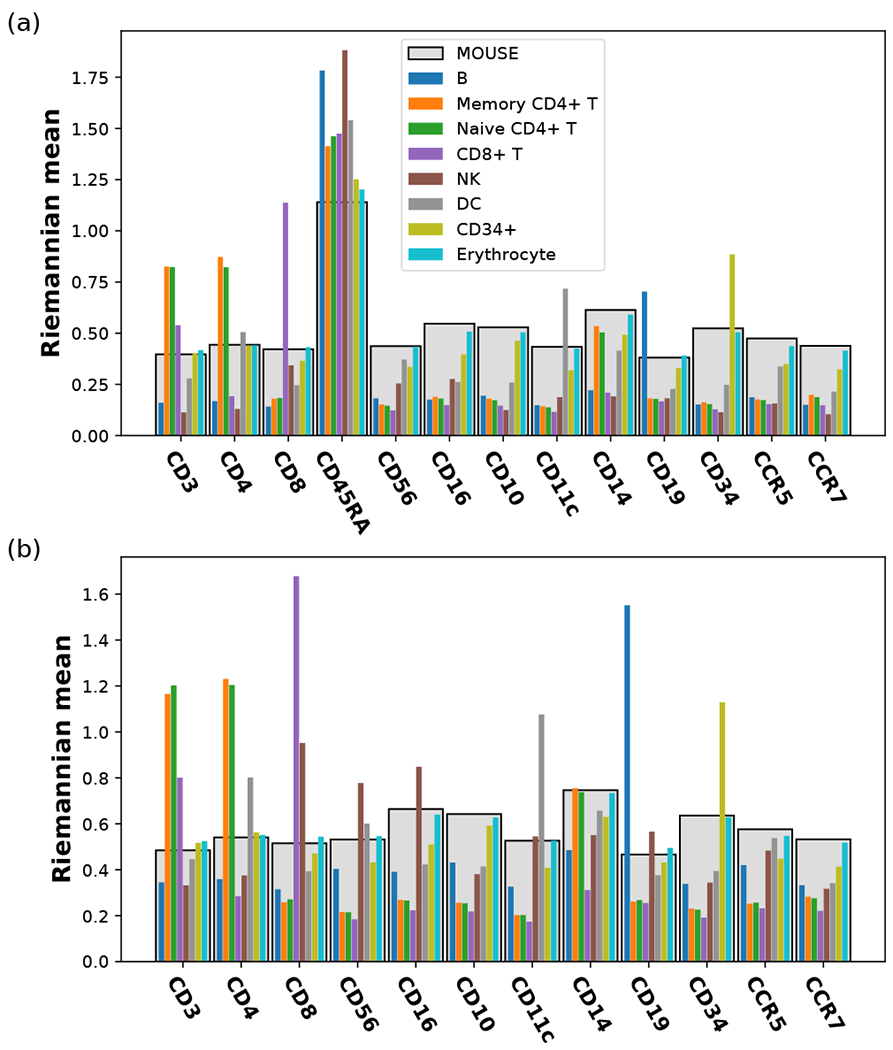
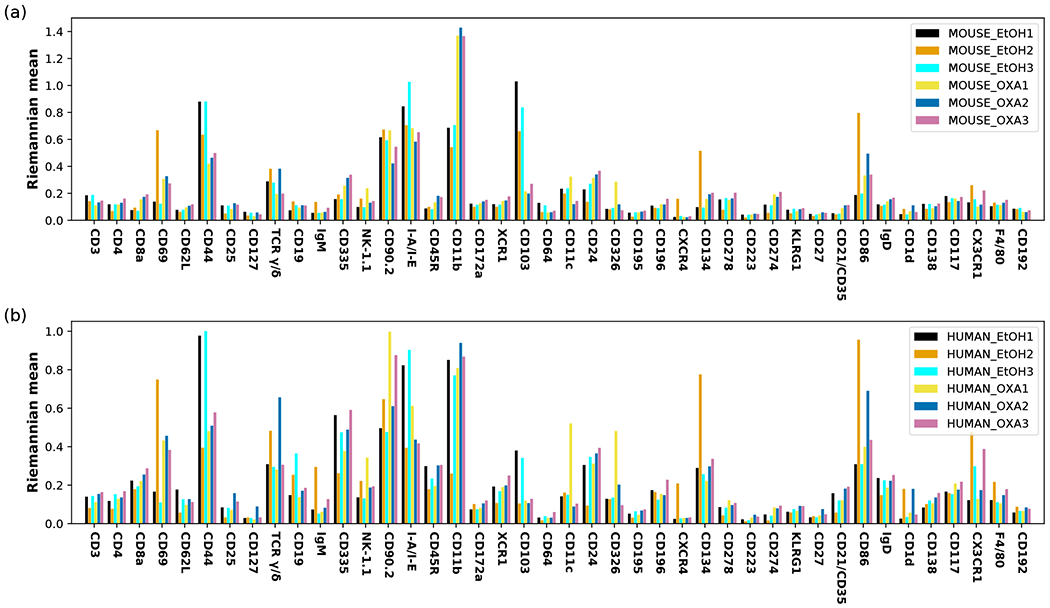
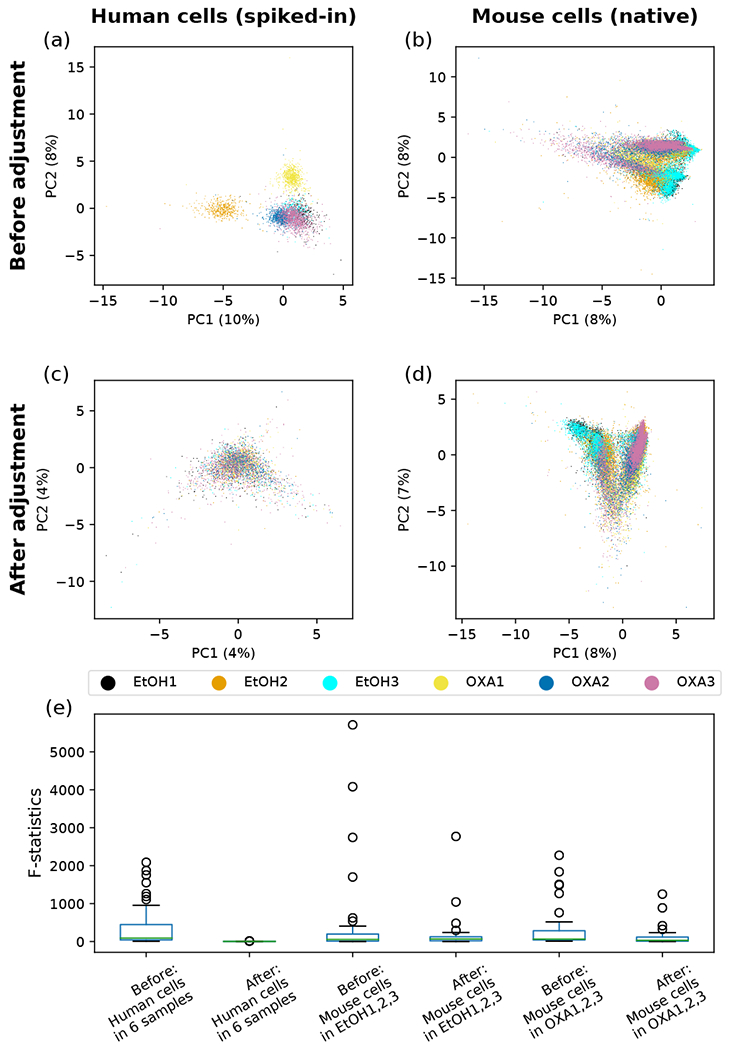
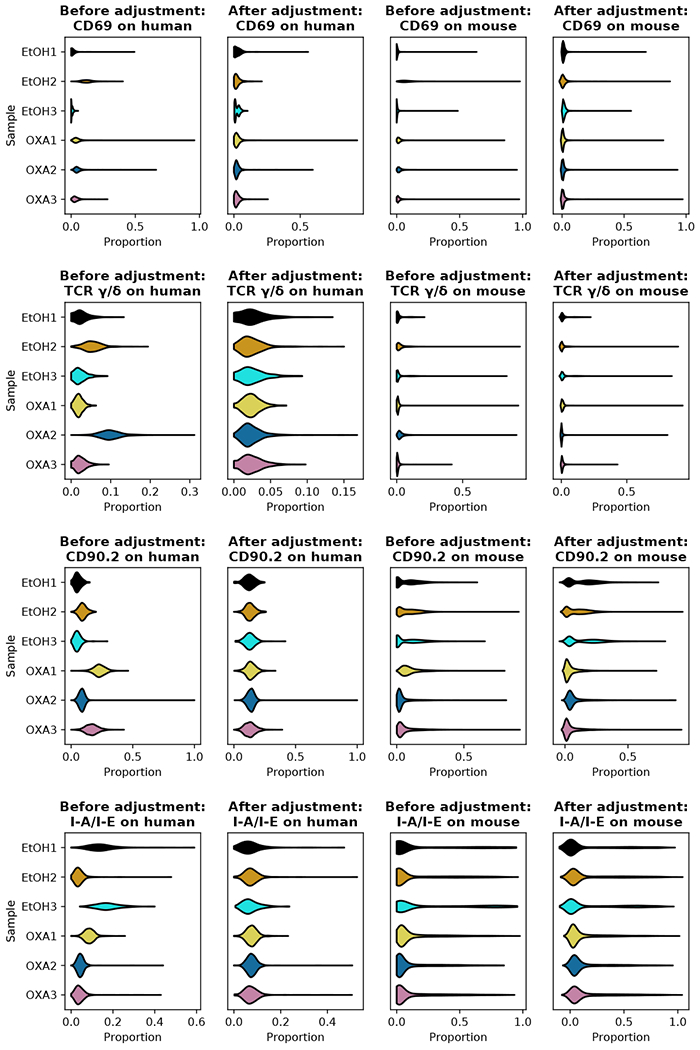
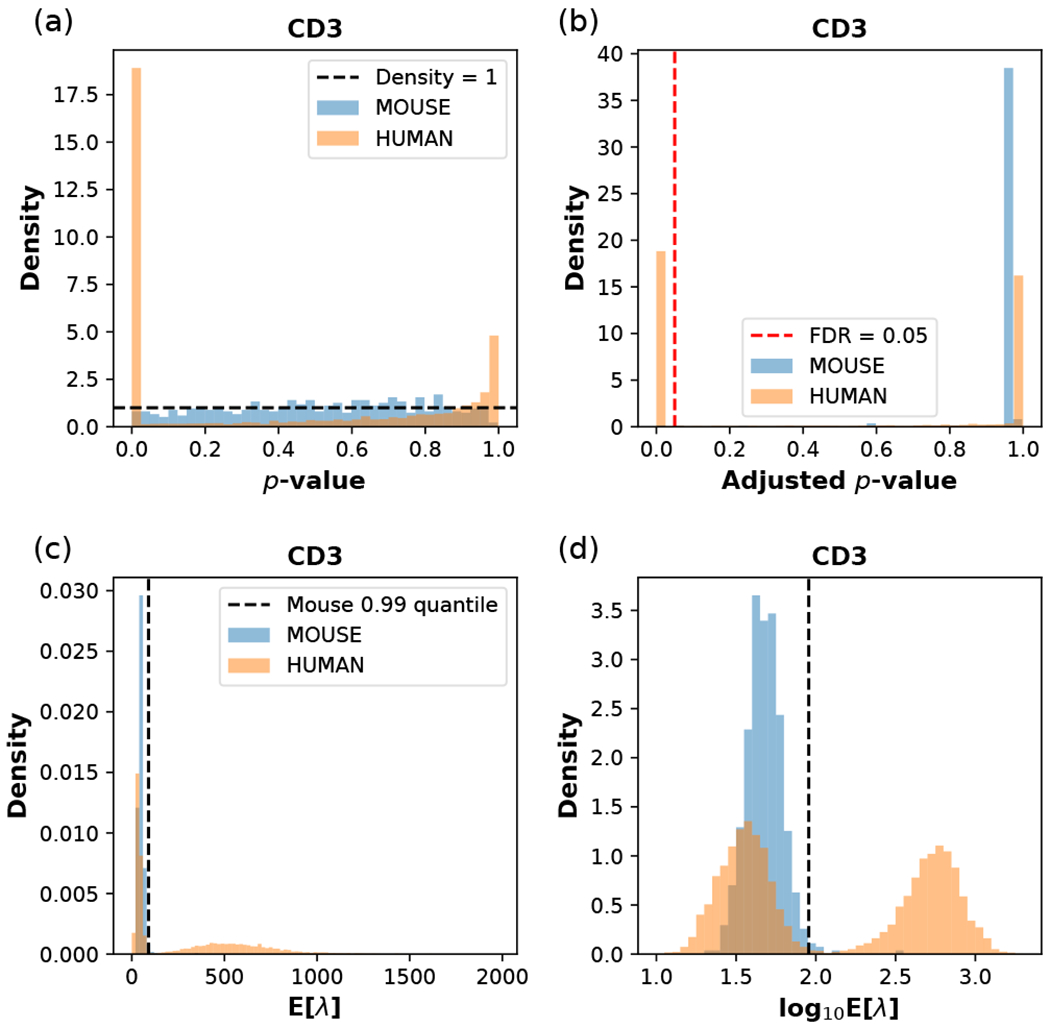
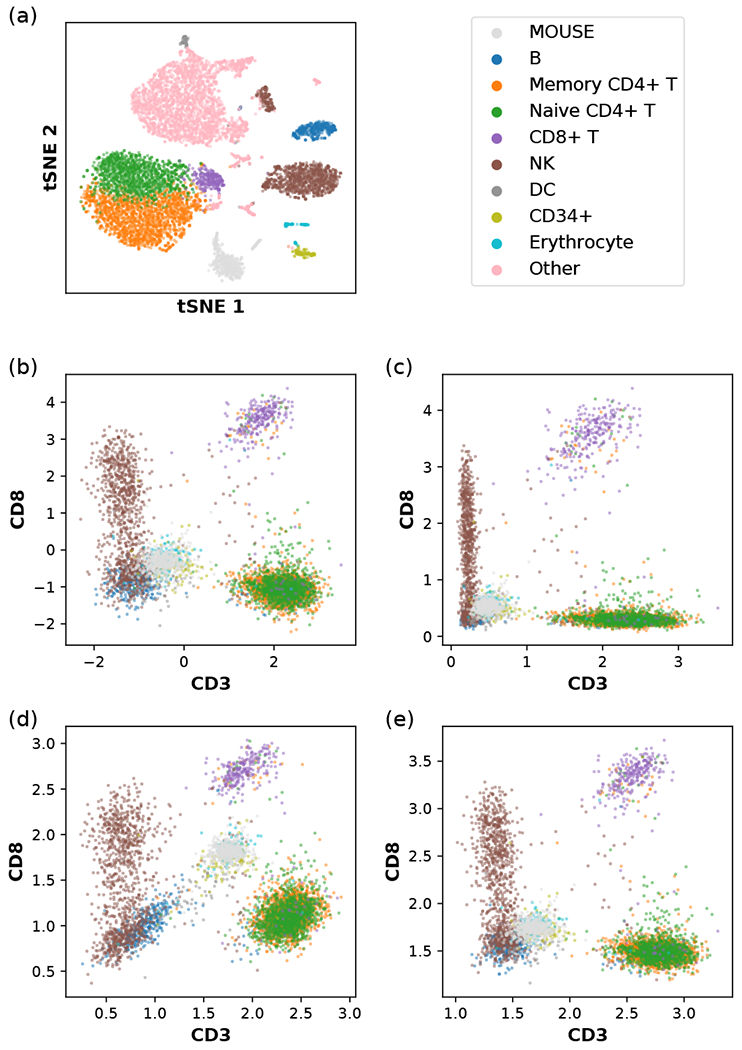
Recent advances in next generation sequencing-based single-cell technologies have allowed high-throughput quantitative detection of cell-surface proteins along with the transcriptome in individual cells, extending our understanding of the heterogeneity of cell populations in diverse tissues that are in different diseased states or under different experimental conditions. Count data of surface proteins from the cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) technology pose new computational challenges, and there is currently a dearth of rigorous mathematical tools for analyzing the data. This work utilizes concepts and ideas from Riemannian geometry to remove batch effects between samples and develops a statistical framework for distinguishing positive signals from background noise. The strengths of these approaches are demonstrated on two independent CITE-seq data sets in mouse and human.
Figures
References
-
- Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, Moore R, McClanahan TK, Sadekova S, and Klappenbach JA, Multiplexed quantification of proteins and transcripts in single cells, Nature Biotechnology 35, 936 (2017). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources