

Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas
- PMID: 32795401
- PMCID: PMC7494613
- DOI: 10.1016/j.ccell.2020.07.008
Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas
Abstract
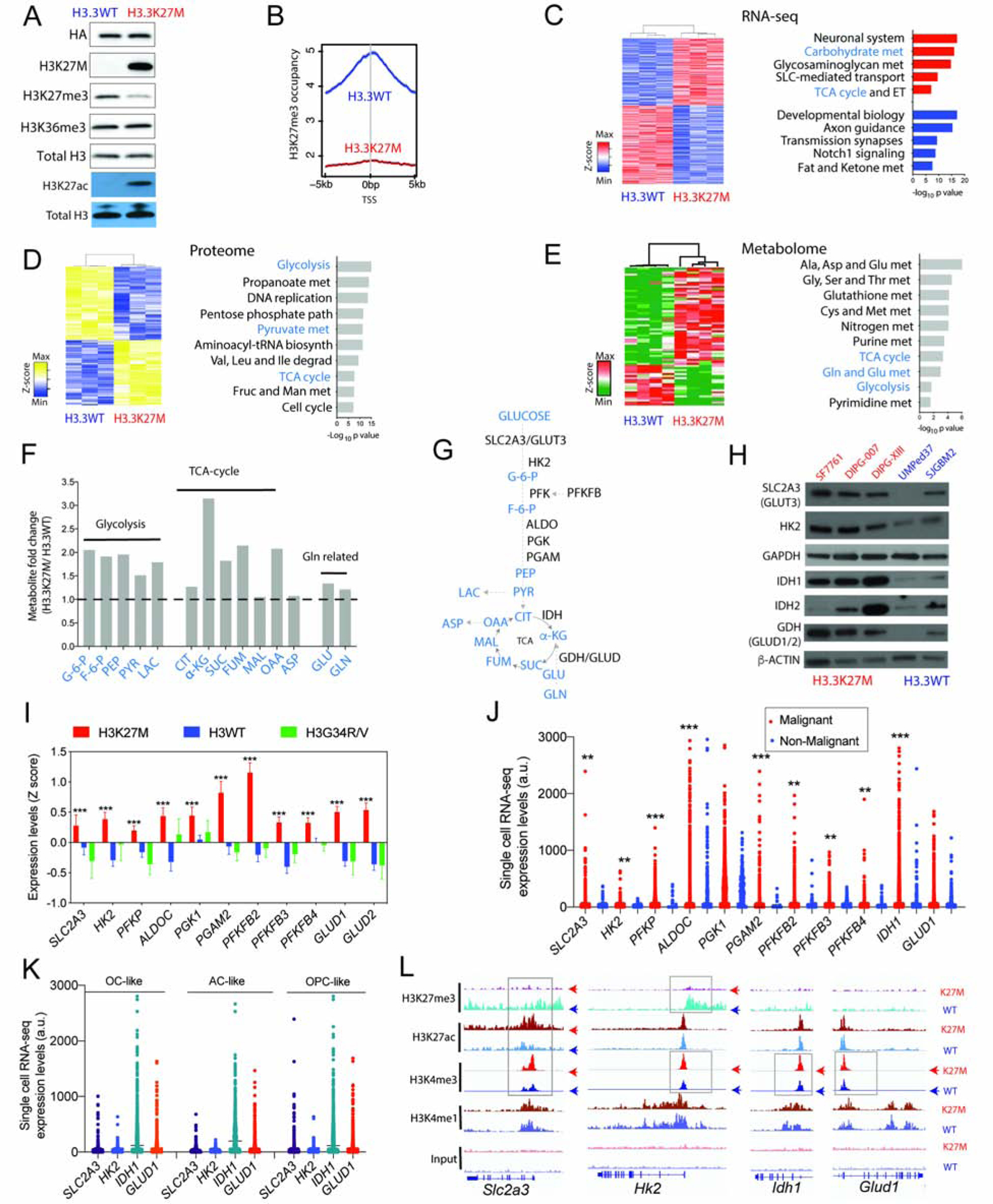
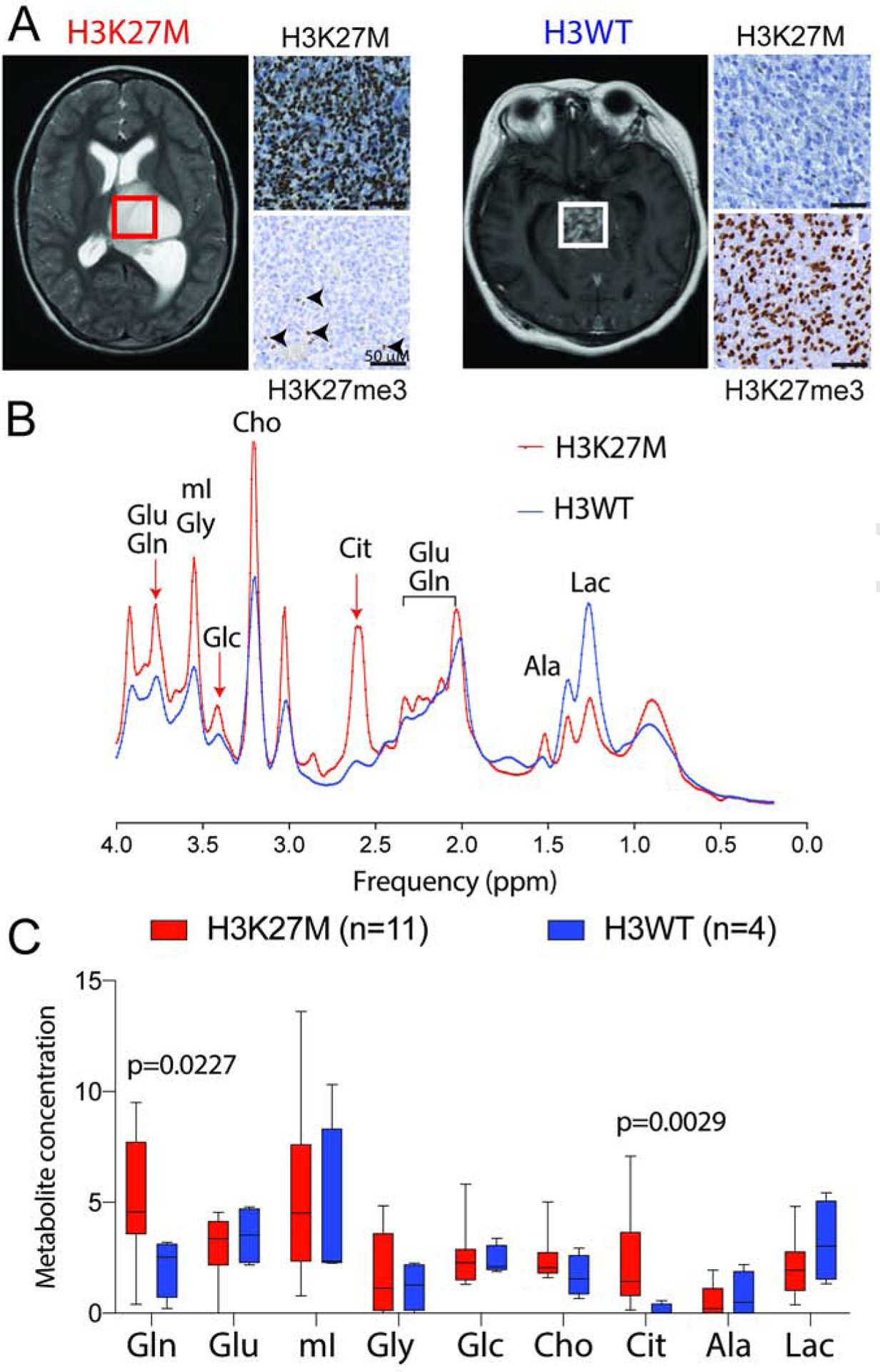
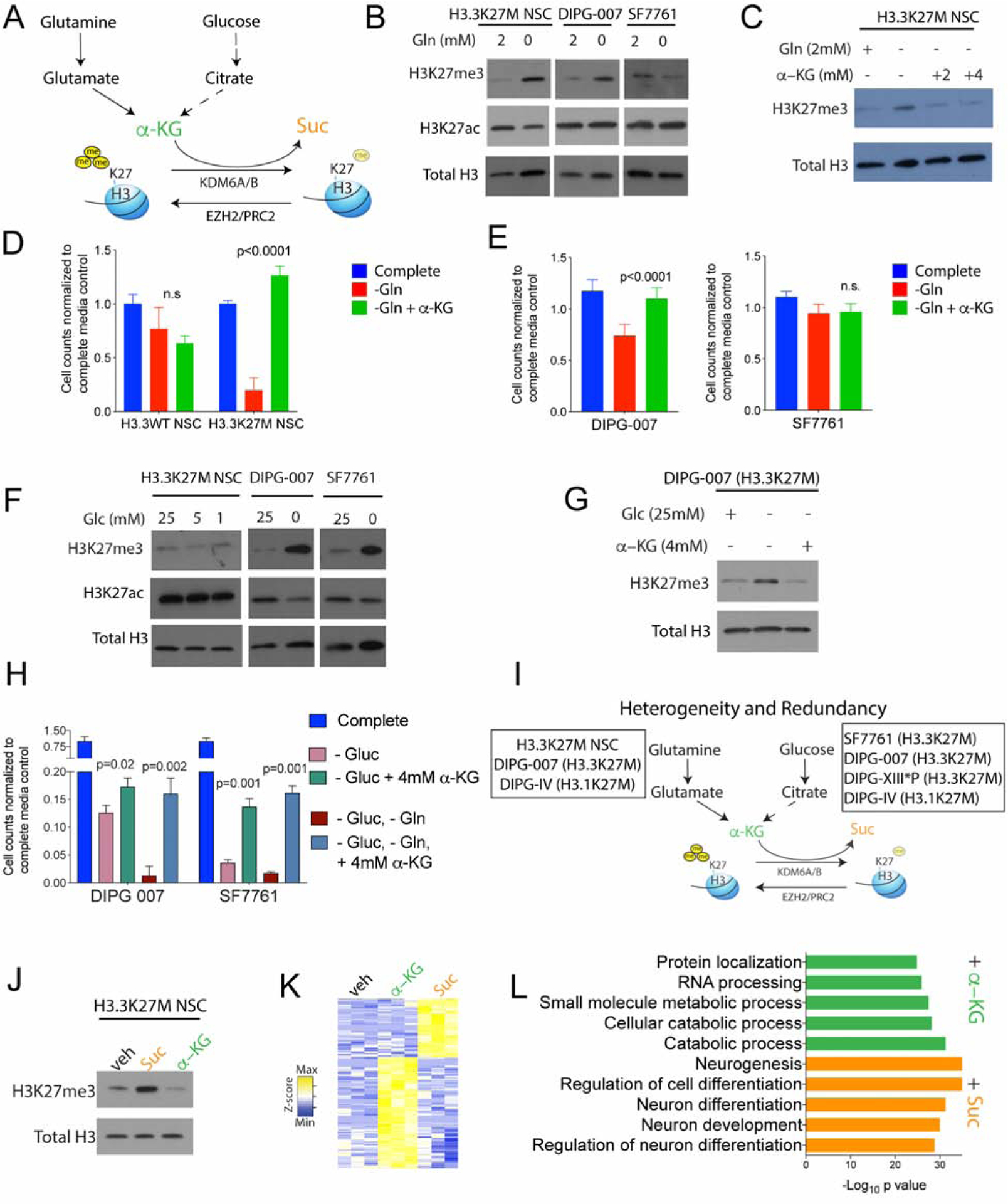
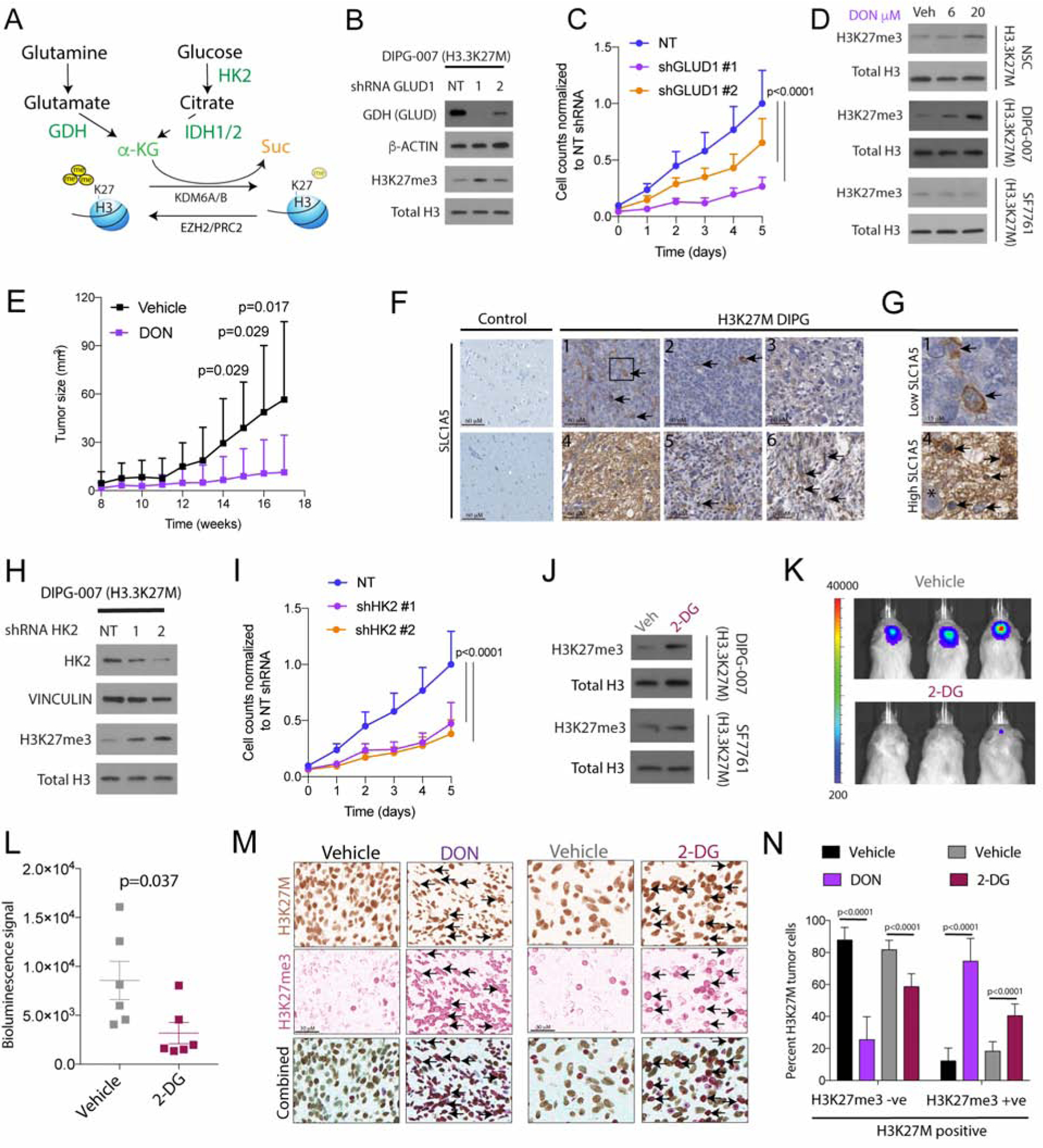
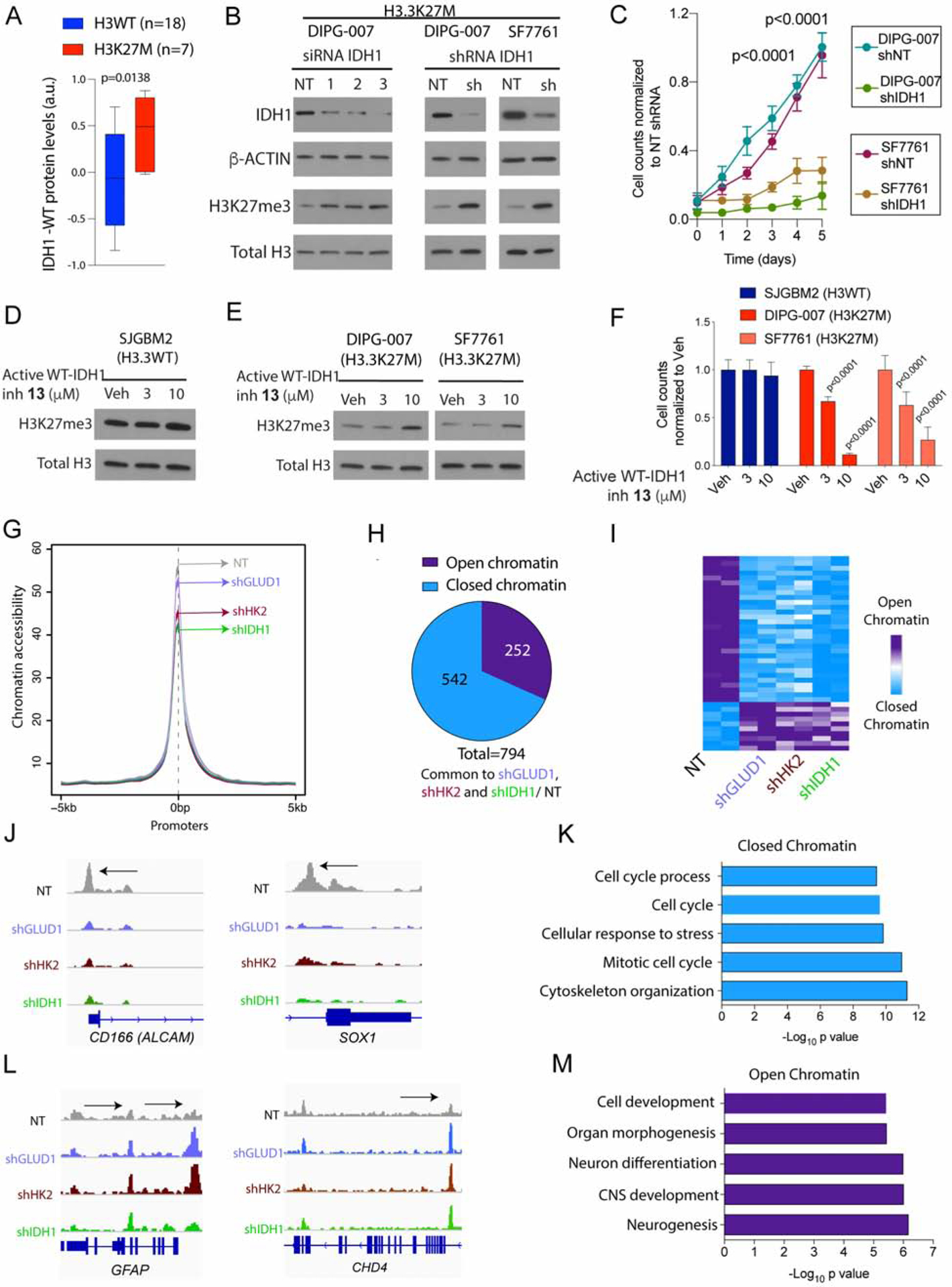
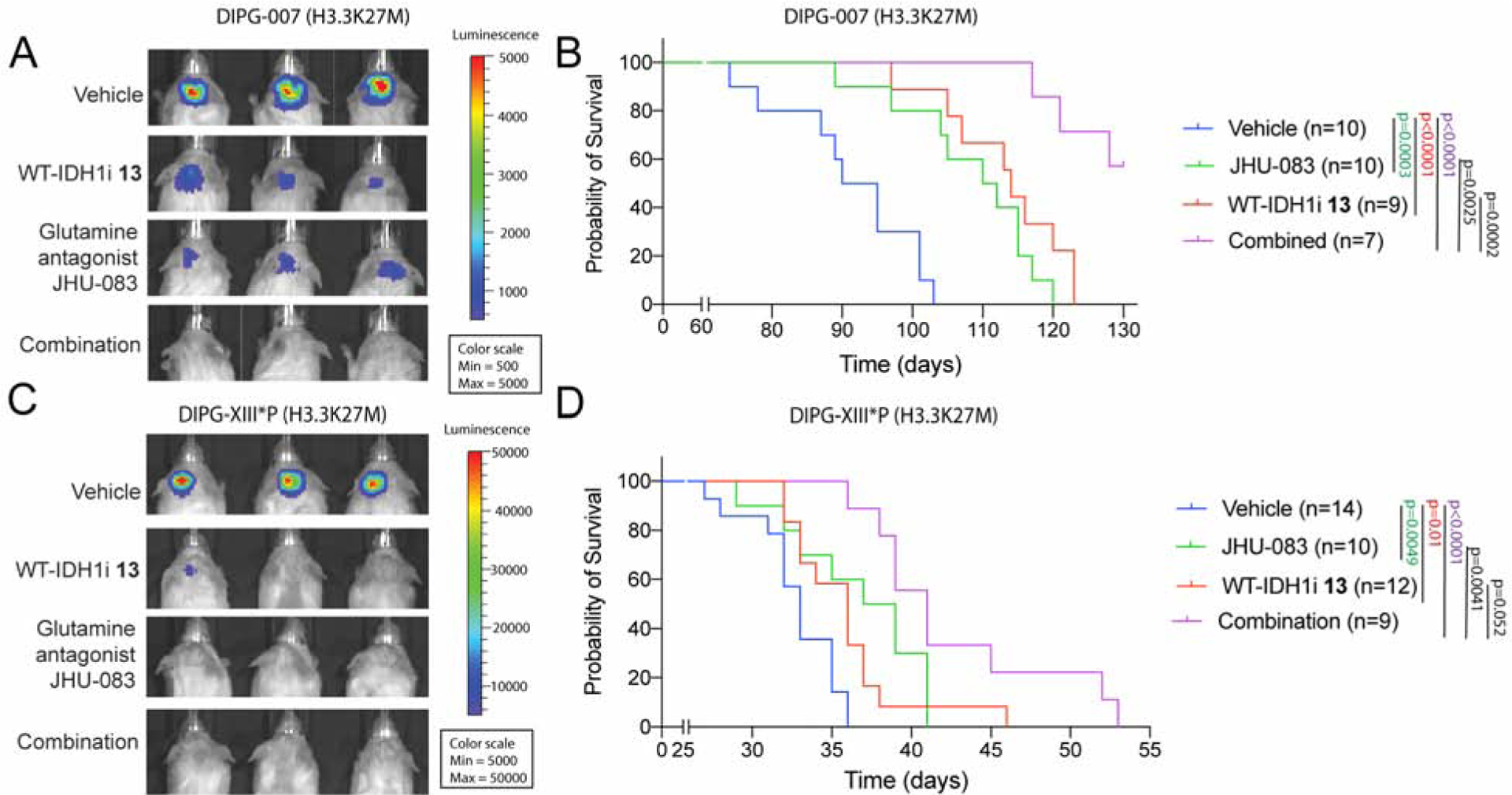
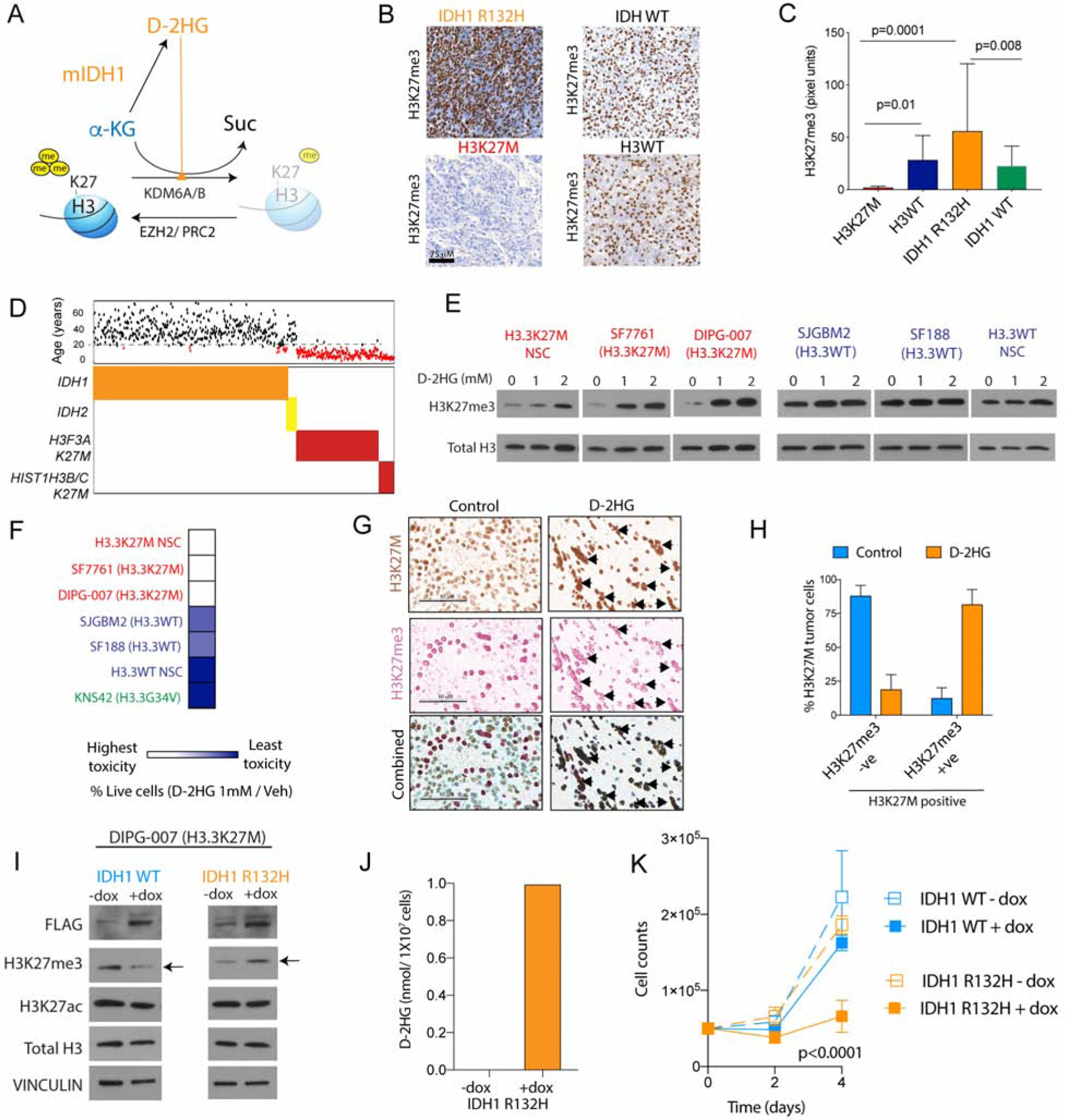
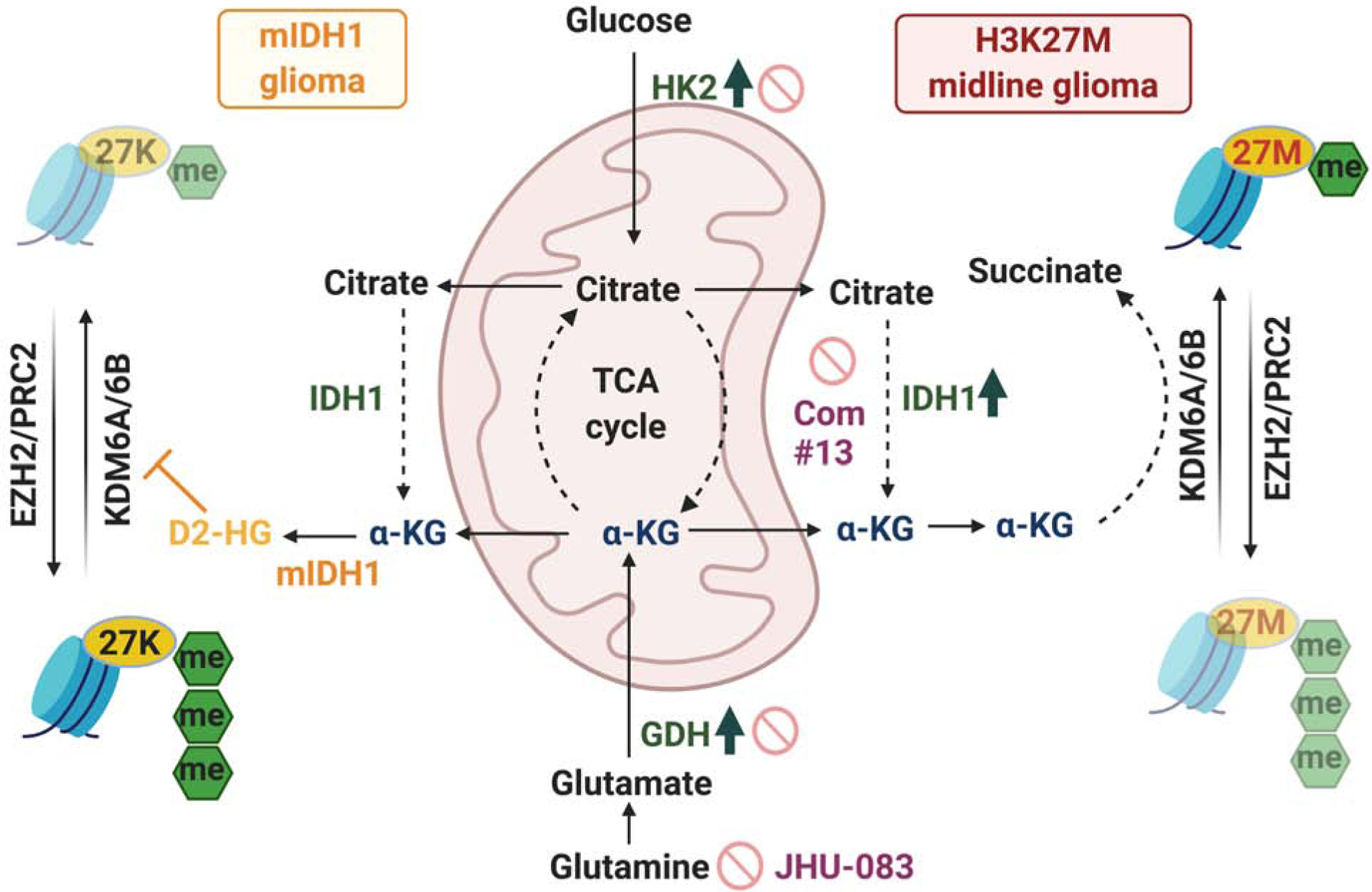
H3K27M diffuse intrinsic pontine gliomas (DIPGs) are fatal and lack treatments. They mainly harbor H3.3K27M mutations resulting in H3K27me3 reduction. Integrated analysis in H3.3K27M cells, tumors, and in vivo imaging in patients showed enhanced glycolysis, glutaminolysis, and tricarboxylic acid cycle metabolism with high alpha-ketoglutarate (α-KG) production. Glucose and/or glutamine-derived α-KG maintained low H3K27me3 in H3.3K27M cells, and inhibition of key enzymes in glycolysis or glutaminolysis increased H3K27me3, altered chromatin accessibility, and prolonged survival in animal models. Previous studies have shown that mutant isocitrate-dehydrogenase (mIDH)1/2 glioma cells convert α-KG to D-2-hydroxyglutarate (D-2HG) to increase H3K27me3. Here, we show that H3K27M and IDH1 mutations are mutually exclusive and experimentally synthetic lethal. Overall, we demonstrate that H3.3K27M and mIDH1 hijack a conserved and critical metabolic pathway in opposing ways to maintain their preferred epigenetic state. Consequently, interruption of this metabolic/epigenetic pathway showed potent efficacy in preclinical models, suggesting key therapeutic targets for much needed treatments.
Keywords: D-2HG; DIPG; H3K27me3; IDH mutation; epigenetics; glutaminolysis; glycolysis; histone methylation; histone mutation; metabolism; α-KG.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests J.B.S. and Z.B. declare the following competing financial interest(s): both are current employees of AbbVie. The small-molecule WT-IDH1 inhibitors were provided by AbbVie after a material transfer agreement. AbbVie participated in the interpretation of inhibitor data, review, and approval of the publication. All other authors declare no competing interests.
Figures
Comment in
-
Targeting metabolic/epigenetic pathways: a potential strategy for cancer therapy in diffuse intrinsic pontine gliomas.Signal Transduct Target Ther. 2020 Oct 6;5(1):226. doi: 10.1038/s41392-020-00344-y. Signal Transduct Target Ther. 2020. PMID: 33024078 Free PMC article. No abstract available.
References
-
- Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, Blanco MA, Giera S, Gillespie SM, Das J, et al. (2019). Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 36, 528–544 e510. - PubMed
-
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
