

The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: A case series
- PMID: 32796131
- PMCID: PMC7713742
- DOI: 10.1212/WNL.0000000000010327
The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: A case series
Abstract
Objective: To clarify the prevalence, long-term natural history, and severity determinants of SEPN1-related myopathy (SEPN1-RM), we analyzed a large international case series.
Methods: Retrospective clinical, histologic, and genetic analysis of 132 pediatric and adult patients (2-58 years) followed up for several decades.
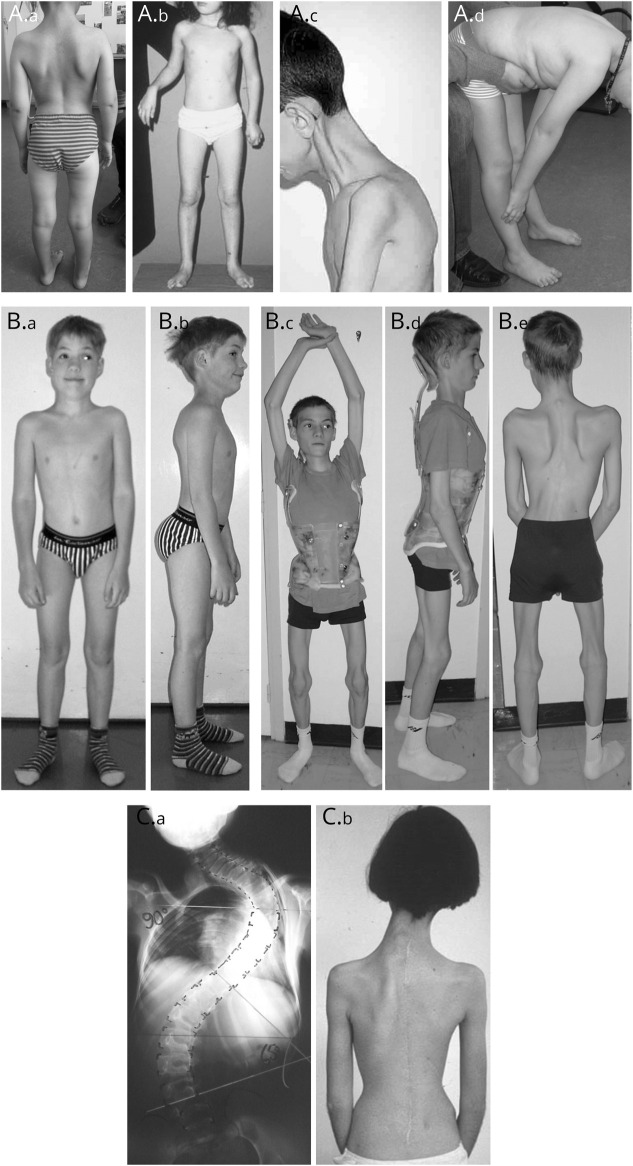
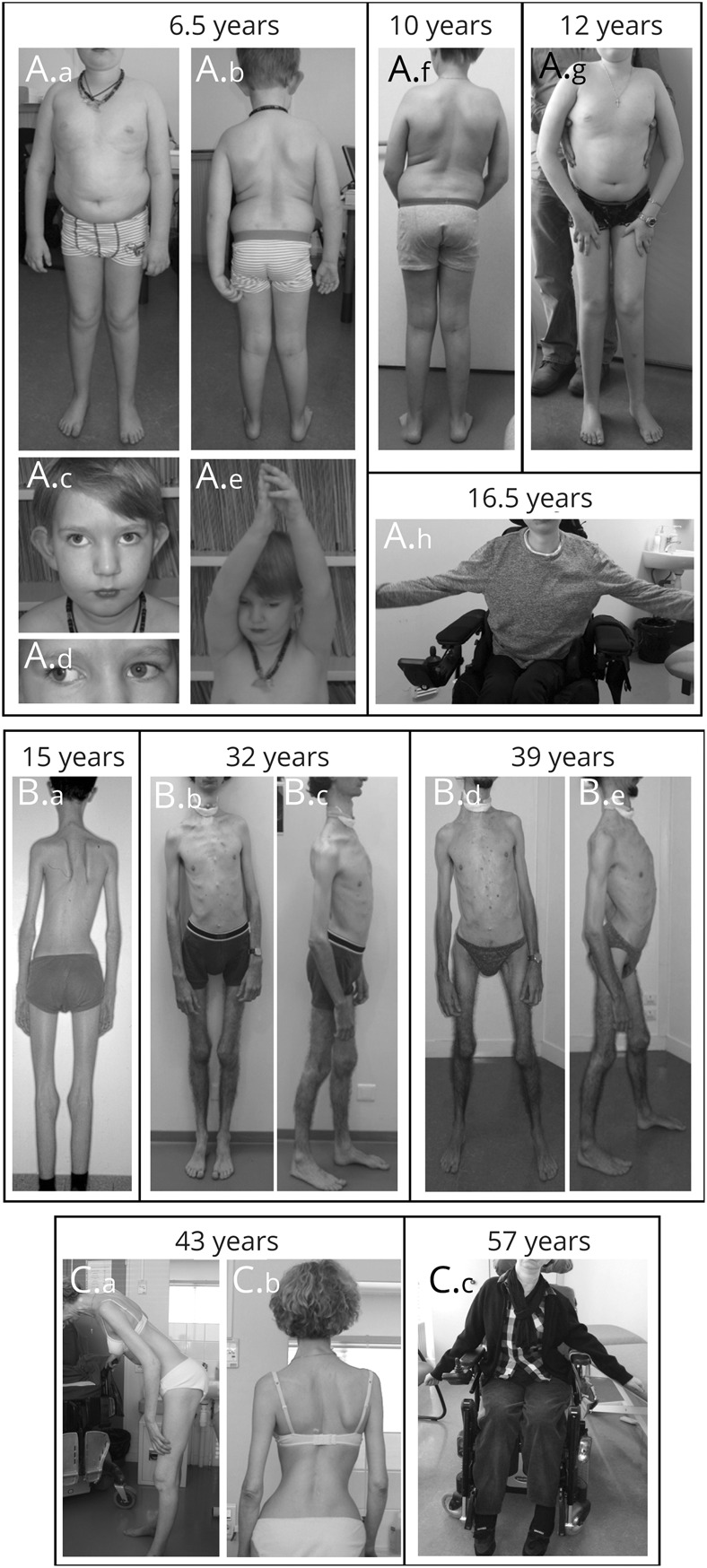
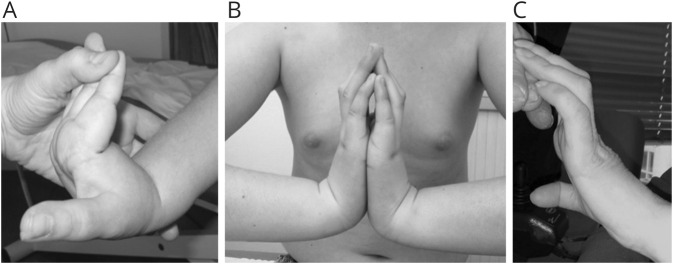
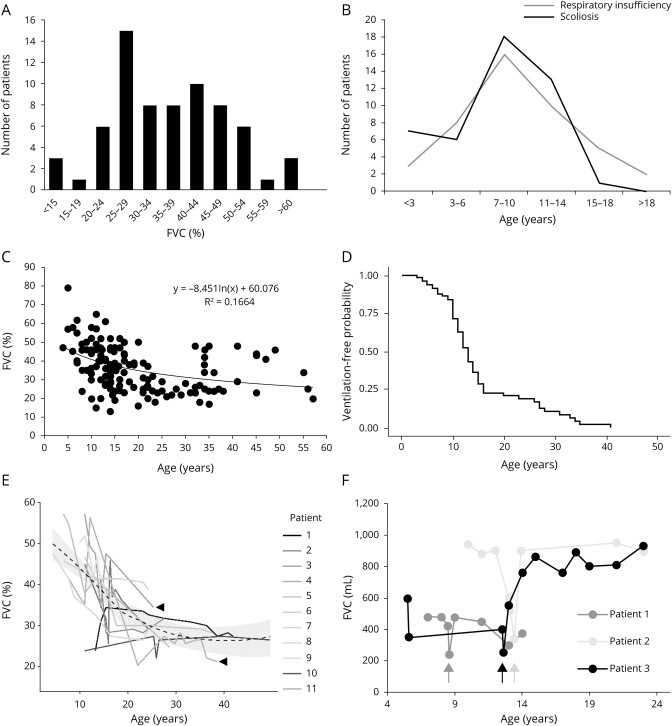
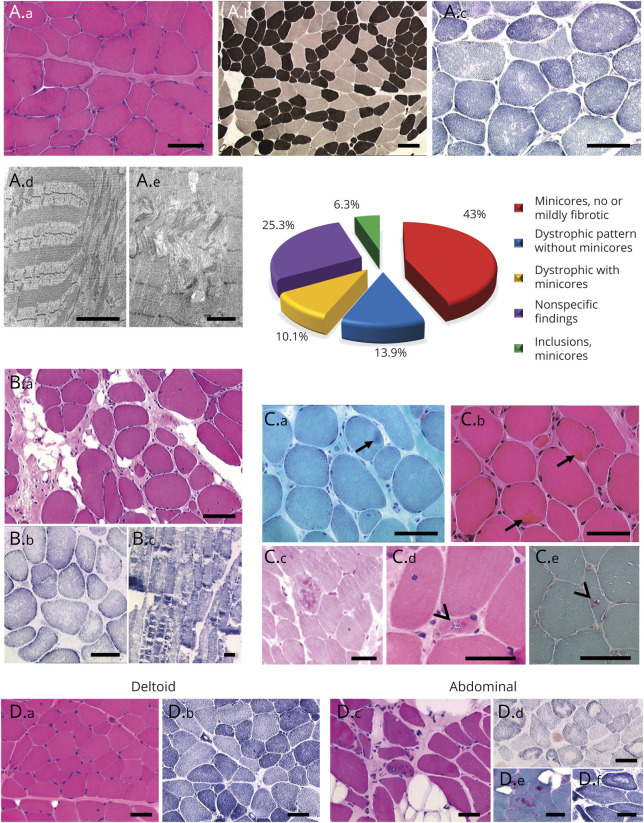
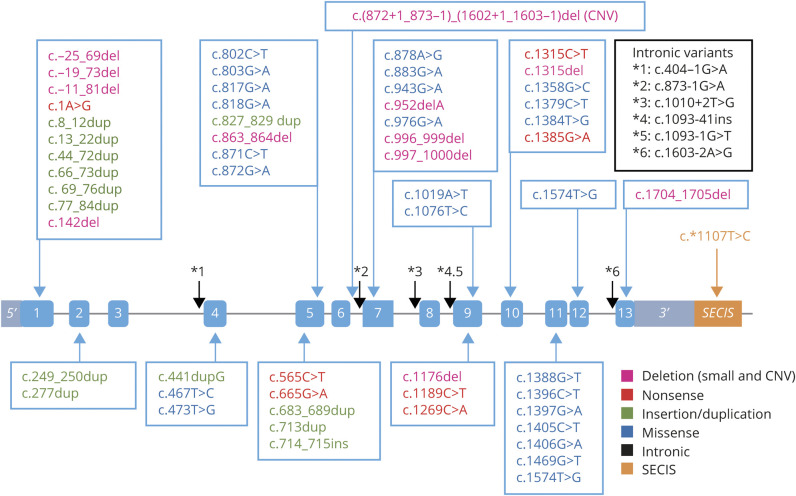
Results: The clinical phenotype was marked by severe axial muscle weakness, spinal rigidity, and scoliosis (86.1%, from 8.9 ± 4 years), with relatively preserved limb strength and previously unreported ophthalmoparesis in severe cases. All patients developed respiratory failure (from 10.1±6 years), 81.7% requiring ventilation while ambulant. Histopathologically, 79 muscle biopsies showed large variability, partly determined by site of biopsy and age. Multi-minicores were the most common lesion (59.5%), often associated with mild dystrophic features and occasionally with eosinophilic inclusions. Identification of 65 SEPN1 mutations, including 32 novel ones and the first pathogenic copy number variation, unveiled exon 1 as the main mutational hotspot and revealed the first genotype-phenotype correlations, bi-allelic null mutations being significantly associated with disease severity (p = 0.017). SEPN1-RM was more severe and progressive than previously thought, leading to loss of ambulation in 10% of cases, systematic functional decline from the end of the third decade, and reduced lifespan even in mild cases. The main prognosis determinants were scoliosis/respiratory management, SEPN1 mutations, and body mass abnormalities, which correlated with disease severity. We propose a set of severity criteria, provide quantitative data for outcome identification, and establish a need for age stratification.
Conclusion: Our results inform clinical practice, improving diagnosis and management, and represent a major breakthrough for clinical trial readiness in this not so rare disease.
© 2020 American Academy of Neurology.
Figures
References
-
- Lescure A, Gautheret D, Carbon P, Krol A. Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. J Biol Chem 1999;274:38147–38154. - PubMed
-
- Moghadaszadeh B, Petit N, Jaillard C, et al. . Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet 2001;29:17–18. - PubMed
-
- Clarke NF, Kidson W, Quijano-Roy S, et al. . SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 2006;59:546–552. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical