

Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review
- PMID: 32796538
- PMCID: PMC7463786
- DOI: 10.3390/jcm9082596
Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review
Abstract
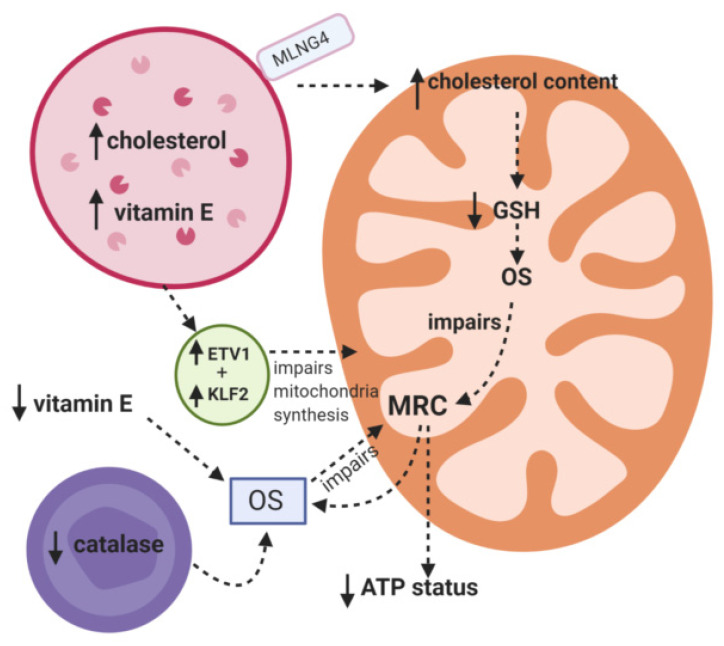
Mitochondrial dysfunction is emerging as an important contributory factor to the pathophysiology of lysosomal storage disorders (LSDs). The cause of mitochondrial dysfunction in LSDs appears to be multifactorial, although impaired mitophagy and oxidative stress appear to be common inhibitory mechanisms shared amongst these heterogeneous disorders. Once impaired, dysfunctional mitochondria may impact upon the function of the lysosome by the generation of reactive oxygen species as well as depriving the lysosome of ATP which is required by the V-ATPase proton pump to maintain the acidity of the lumen. Given the reported evidence of mitochondrial dysfunction in LSDs together with the important symbiotic relationship between these two organelles, therapeutic strategies targeting both lysosome and mitochondrial dysfunction may be an important consideration in the treatment of LSDs. In this review we examine the putative mechanisms that may be responsible for mitochondrial dysfunction in reported LSDs which will be supplemented with morphological and clinical information.
Keywords: autophagy; inflammation; lysosomal storage diseases; mitochondrial dysfunction; mitophagy and cytokine; oxidative stress; reactive oxygen species.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
Publication types
LinkOut - more resources
Full Text Sources
