

Maximizing transcription of nucleic acids with efficient T7 promoters
- PMID: 32796901
- PMCID: PMC7429497
- DOI: 10.1038/s42003-020-01167-x
Maximizing transcription of nucleic acids with efficient T7 promoters
Abstract
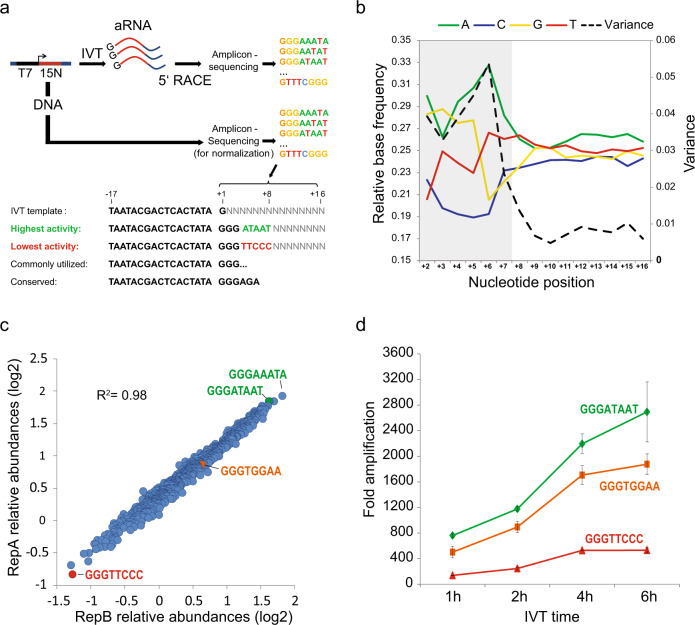
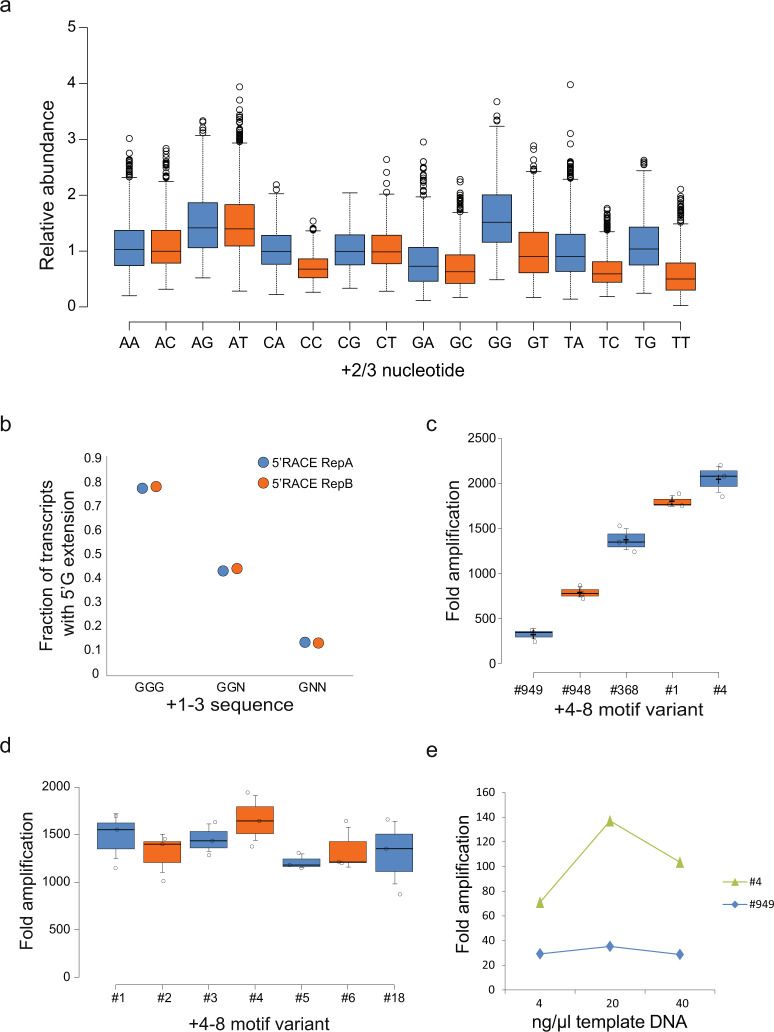
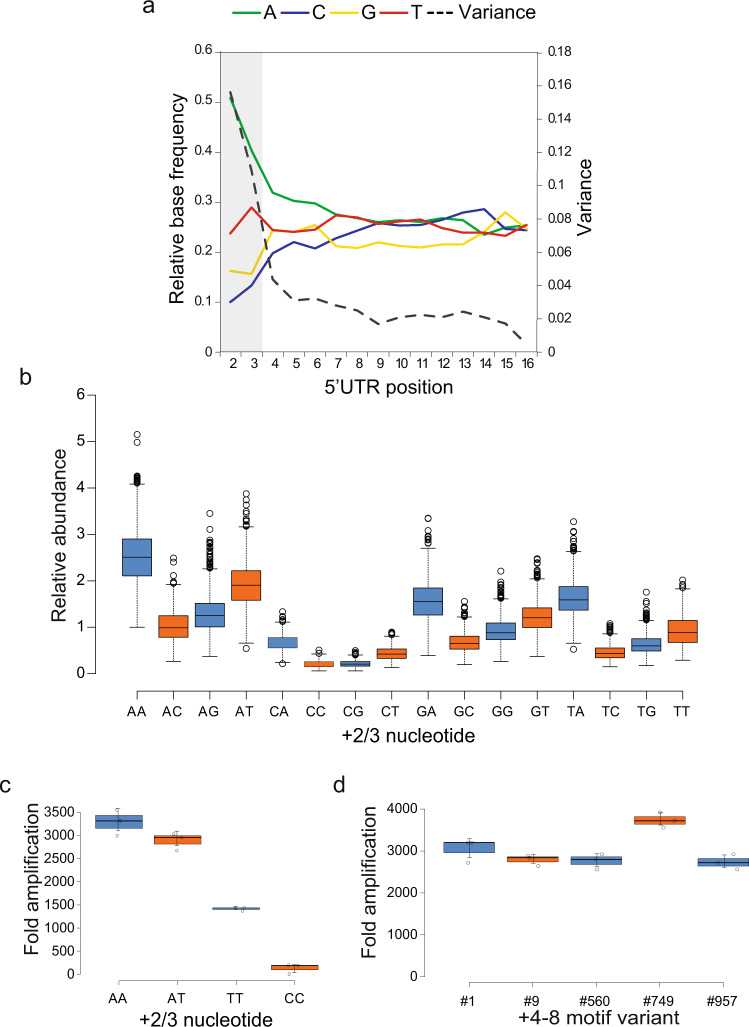
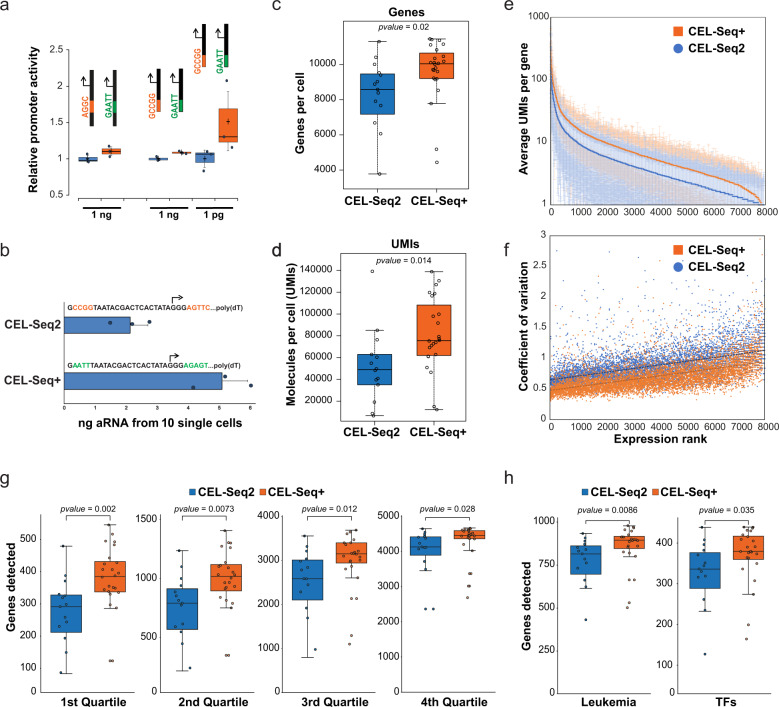
In vitro transcription using T7 bacteriophage polymerase is widely used in molecular biology. Here, we use 5'RACE-Seq to screen a randomized initially transcribed region of the T7 promoter for cross-talk with transcriptional activity. We reveal that sequences from position +4 to +8 downstream of the transcription start site affect T7 promoter activity over a 5-fold range, and identify promoter variants with significantly enhanced transcriptional output that increase the yield of in vitro transcription reactions across a wide range of template concentrations. We furthermore introduce CEL-Seq+ , which uses an optimized T7 promoter to amplify cDNA for single-cell RNA-Sequencing. CEL-Seq+ facilitates scRNA-Seq library preparation, and substantially increases library complexity and the number of expressed genes detected per cell, highlighting a particular value of optimized T7 promoters in bioanalytical applications.
Conflict of interest statement
A patent dealing with optimized T7 and other promoter sequences has been filed by Thomas Conrad and Sascha Sauer (EP19199289). The remaining authors (Izabela Plumbom, Maria Alcobendas, and Ramon Vidal) declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
