

HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads
- PMID: 32801147
- PMCID: PMC7545148
- DOI: 10.1101/gr.263566.120
HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads
Abstract
Complete and accurate genome assemblies form the basis of most downstream genomic analyses and are of critical importance. Recent genome assembly projects have relied on a combination of noisy long-read sequencing and accurate short-read sequencing, with the former offering greater assembly continuity and the latter providing higher consensus accuracy. The recently introduced Pacific Biosciences (PacBio) HiFi sequencing technology bridges this divide by delivering long reads (>10 kbp) with high per-base accuracy (>99.9%). Here we present HiCanu, a modification of the Canu assembler designed to leverage the full potential of HiFi reads via homopolymer compression, overlap-based error correction, and aggressive false overlap filtering. We benchmark HiCanu with a focus on the recovery of haplotype diversity, major histocompatibility complex (MHC) variants, satellite DNAs, and segmental duplications. For diploid human genomes sequenced to 30× HiFi coverage, HiCanu achieved superior accuracy and allele recovery compared to the current state of the art. On the effectively haploid CHM13 human cell line, HiCanu achieved an NG50 contig size of 77 Mbp with a per-base consensus accuracy of 99.999% (QV50), surpassing recent assemblies of high-coverage, ultralong Oxford Nanopore Technologies (ONT) reads in terms of both accuracy and continuity. This HiCanu assembly correctly resolves 337 out of 341 validation BACs sampled from known segmental duplications and provides the first preliminary assemblies of nine complete human centromeric regions. Although gaps and errors still remain within the most challenging regions of the genome, these results represent a significant advance toward the complete assembly of human genomes.
© 2020 Nurk et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
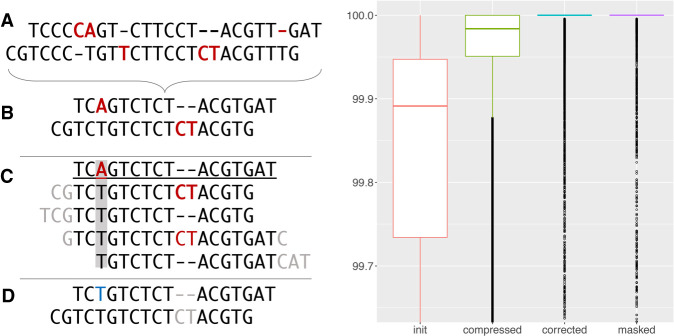
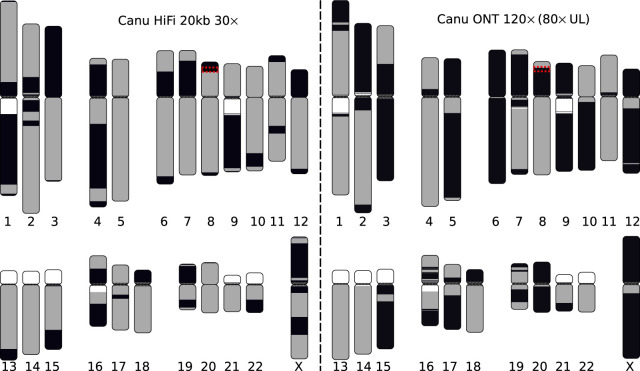
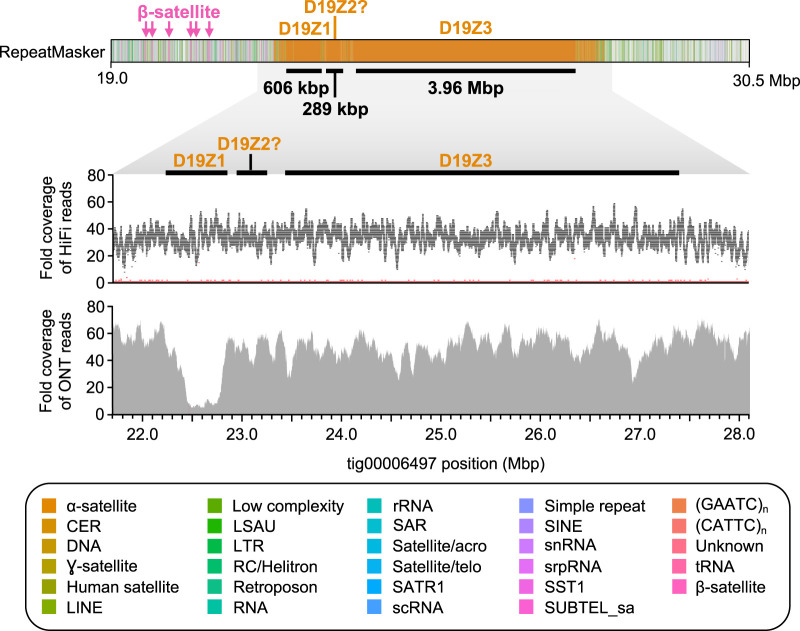
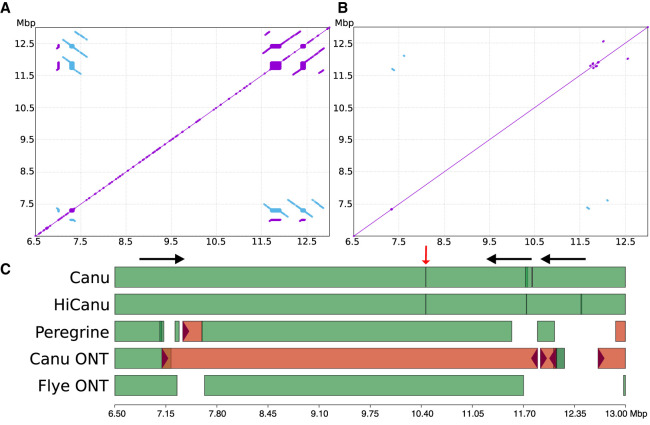
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous