

Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression
- PMID: 32801214
- PMCID: PMC7474612
- DOI: 10.1073/pnas.2007412117
Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression
Abstract
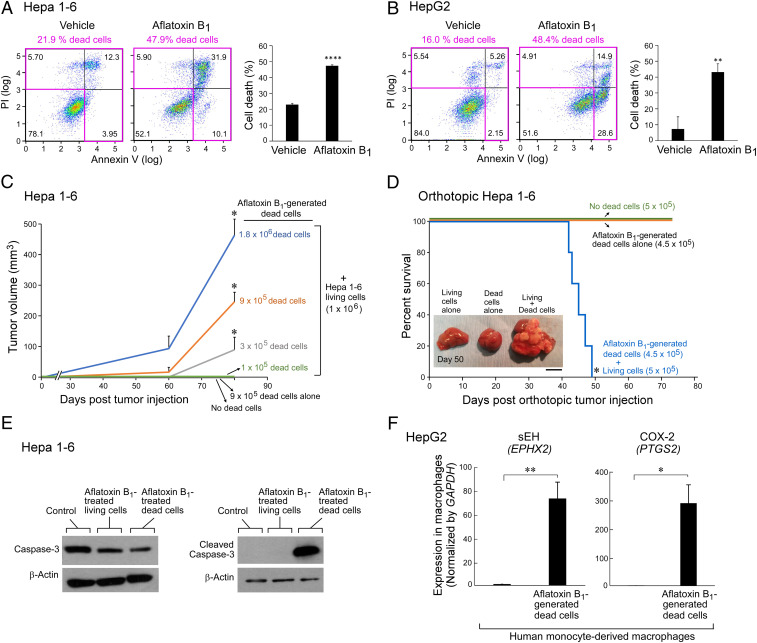
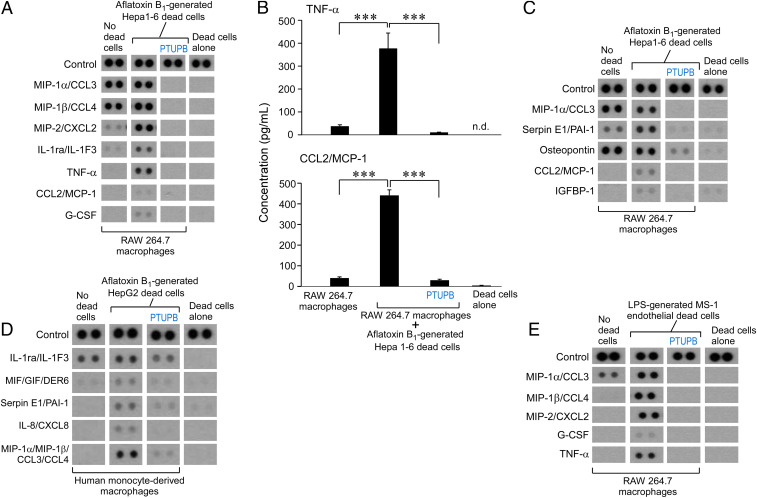
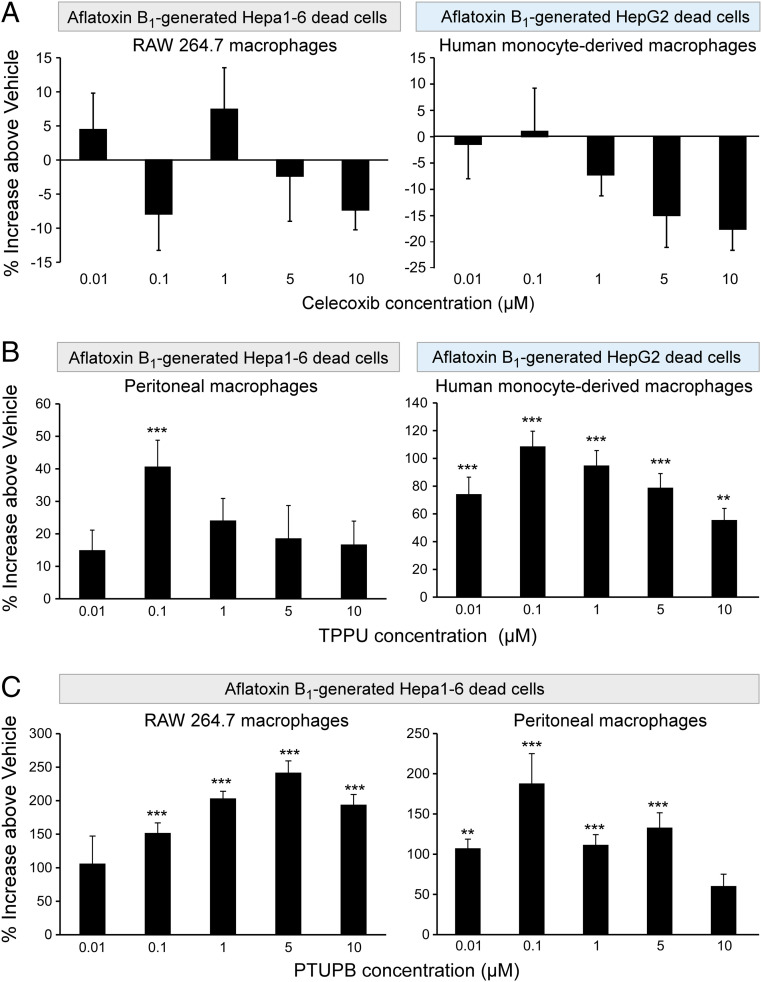
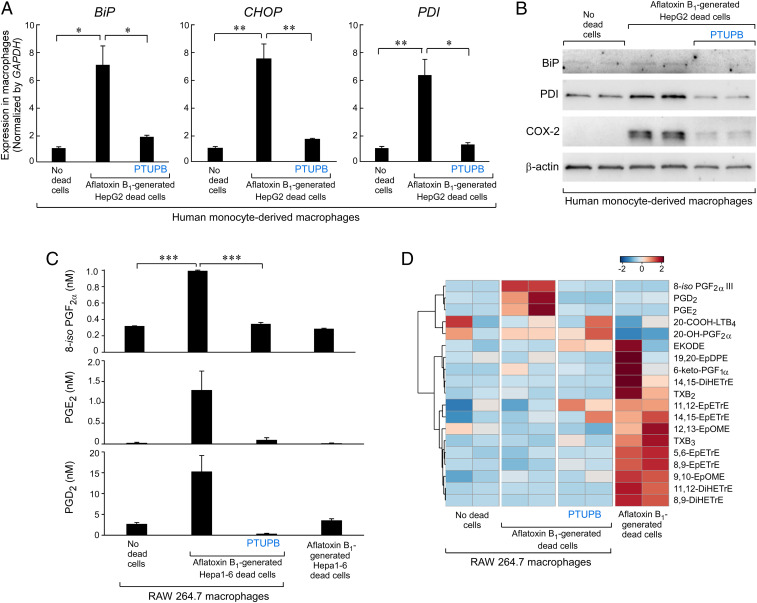
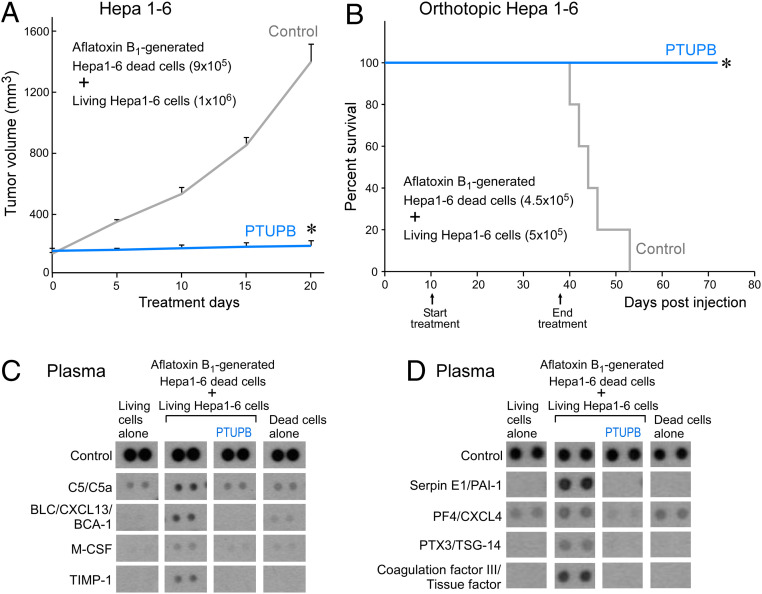
Toxic environmental carcinogens promote cancer via genotoxic and nongenotoxic pathways, but nongenetic mechanisms remain poorly characterized. Carcinogen-induced apoptosis may trigger escape from dormancy of microtumors by interfering with inflammation resolution and triggering an endoplasmic reticulum (ER) stress response. While eicosanoid and cytokine storms are well-characterized in infection and inflammation, they are poorly characterized in cancer. Here, we demonstrate that carcinogens, such as aflatoxin B1 (AFB1), induce apoptotic cell death and the resulting cell debris stimulates hepatocellular carcinoma (HCC) tumor growth via an "eicosanoid and cytokine storm." AFB1-generated debris up-regulates cyclooxygenase-2 (COX-2), soluble epoxide hydrolase (sEH), ER stress-response genes including BiP, CHOP, and PDI in macrophages. Thus, selective cytokine or eicosanoid blockade is unlikely to prevent carcinogen-induced cancer progression. Pharmacological abrogation of both the COX-2 and sEH pathways by PTUPB prevented the debris-stimulated eicosanoid and cytokine storm, down-regulated ER stress genes, and promoted macrophage phagocytosis of debris, resulting in suppression of HCC tumor growth. Thus, inflammation resolution via dual COX-2/sEH inhibition is an approach to prevent carcinogen-induced cancer.
Keywords: bioactive lipid; carcinogenesis; cell death; inflammation resolution; soluble epoxide hydrolase.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Kensler T. W. et al. ., Predictive value of molecular dosimetry: Individual versus group effects of oltipraz on aflatoxin-albumin adducts and risk of liver cancer. Cancer Epidemiol. Biomarkers Prev. 6, 603–610 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
