

Integrated sequencing and array comparative genomic hybridization in familial Parkinson disease
- PMID: 32802956
- PMCID: PMC7413630
- DOI: 10.1212/NXG.0000000000000498
Integrated sequencing and array comparative genomic hybridization in familial Parkinson disease
Abstract
Objective: To determine how single nucleotide variants (SNVs) and copy number variants (CNVs) contribute to molecular diagnosis in familial Parkinson disease (PD), we integrated exome sequencing (ES) and genome-wide array-based comparative genomic hybridization (aCGH) and further probed CNV structure to reveal mutational mechanisms.
Methods: We performed ES on 110 subjects with PD and a positive family history; 99 subjects were also evaluated using genome-wide aCGH. We interrogated ES and aCGH data for pathogenic SNVs and CNVs at Mendelian PD gene loci. We confirmed SNVs via Sanger sequencing and further characterized CNVs with custom-designed high-density aCGH, droplet digital PCR, and breakpoint sequencing.
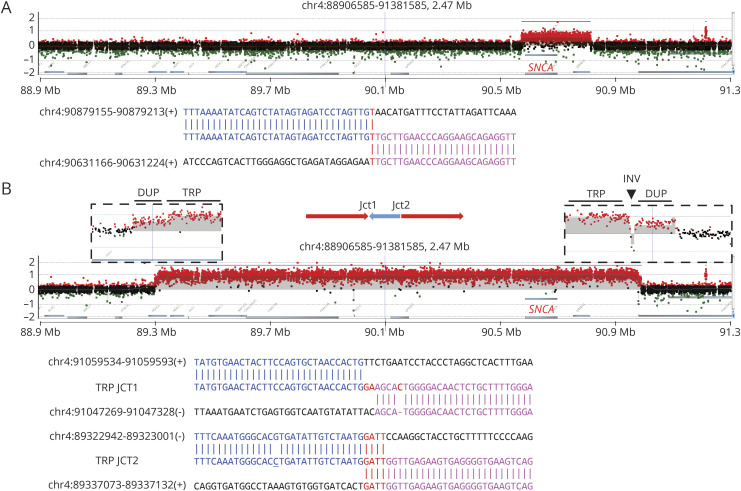
Results: Using ES, we discovered individuals with known pathogenic SNVs in GBA (p.Glu365Lys, p.Thr408Met, p.Asn409Ser, and p.Leu483Pro) and LRRK2 (p.Arg1441Gly and p.Gly2019Ser). Two subjects were each double heterozygotes for variants in GBA and LRRK2. Based on aCGH, we additionally discovered cases with an SNCA duplication and heterozygous intragenic GBA deletion. Five additional subjects harbored both SNVs (p.Asn52Metfs*29, p.Thr240Met, p.Pro437Leu, and p.Trp453*) and likely disrupting CNVs at the PRKN locus, consistent with compound heterozygosity. In nearly all cases, breakpoint sequencing revealed microhomology, a mutational signature consistent with CNV formation due to DNA replication errors.
Conclusions: Integrated ES and aCGH yielded a genetic diagnosis in 19.3% of our familial PD cohort. Our analyses highlight potential mechanisms for SNCA and PRKN CNV formation, uncover multilocus pathogenic variation, and identify novel SNVs and CNVs for further investigation as potential PD risk alleles.
Copyright © 2020 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures



References
-
- Elbaz A, Grigoletto F, Baldereschi M, et al. Familial aggregation of Parkinson's disease: a population-based case-control study in Europe. EUROPARKINSON Study Group. Neurology 1999;52:1876–1882. - PubMed
-
- Espay AJ, Brundin P, Lang AE. Precision medicine for disease modification in Parkinson disease. Nat Rev Neurol 2017;13:119–126. - PubMed
-
- Brás J, Guerreiro R, Hardy J. SnapShot: genetics of Parkinson's disease. Cell 2015;160:570–570.e1. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous