

The T cell differentiation landscape is shaped by tumour mutations in lung cancer
- PMID: 32803172
- PMCID: PMC7115931
- DOI: 10.1038/s43018-020-0066-y
The T cell differentiation landscape is shaped by tumour mutations in lung cancer
Abstract
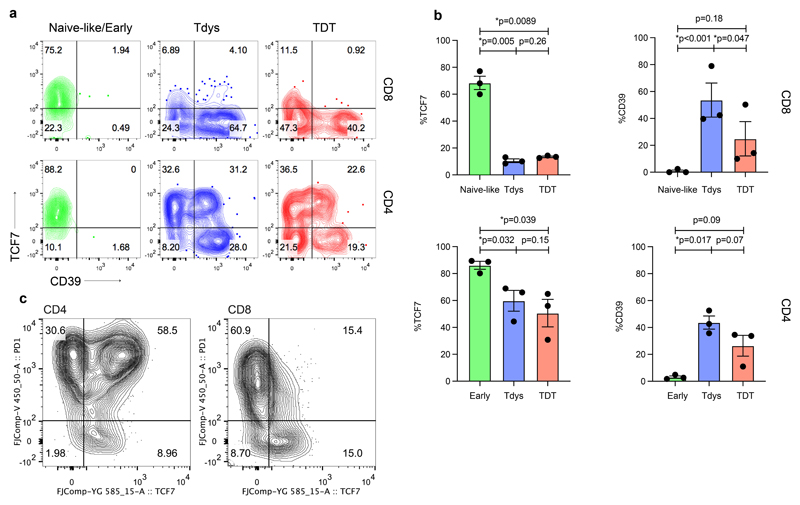
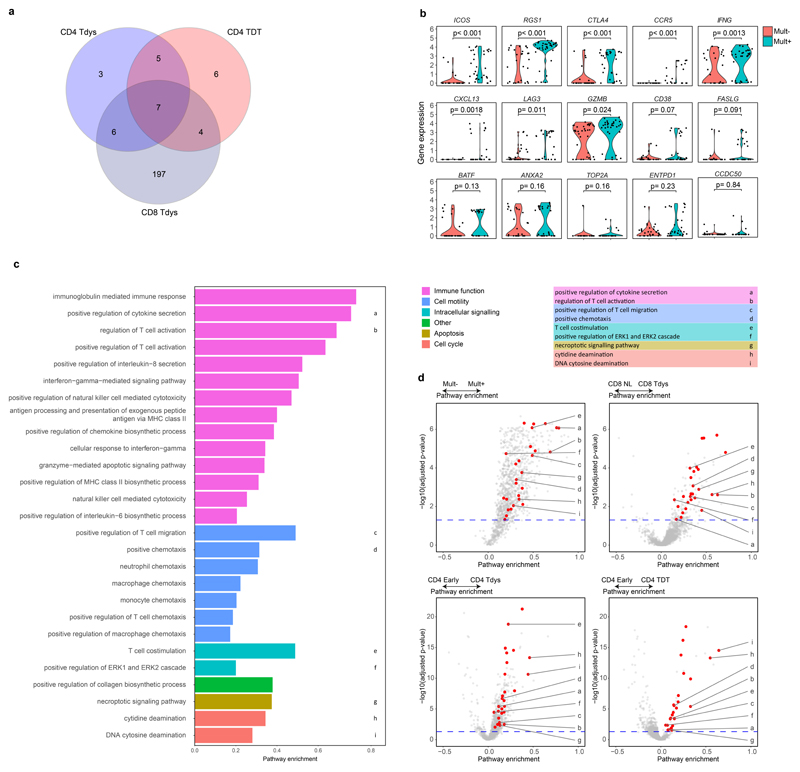
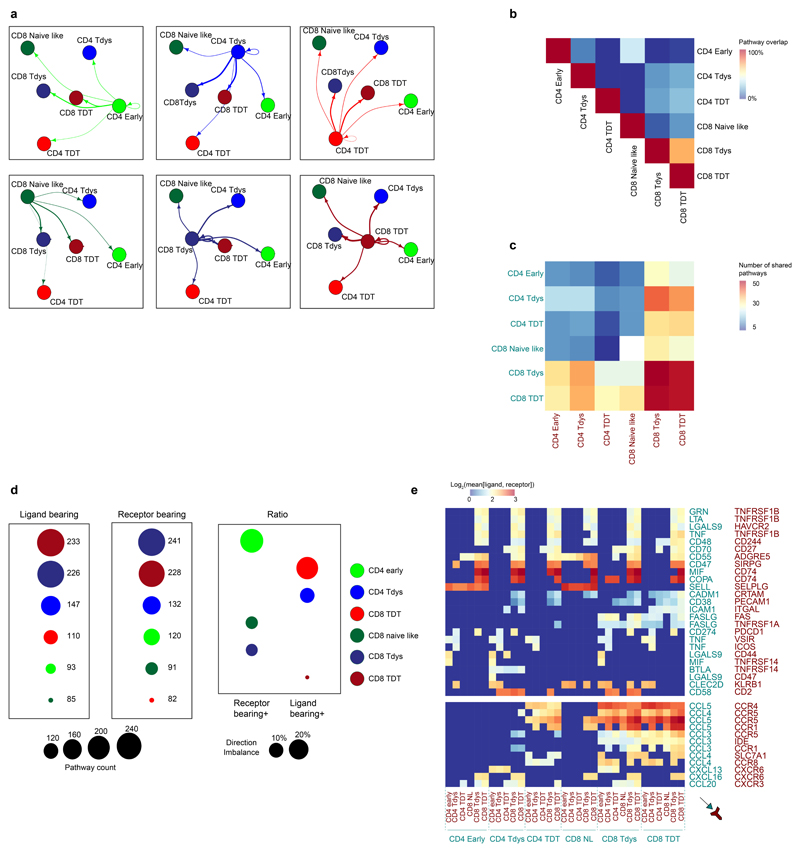
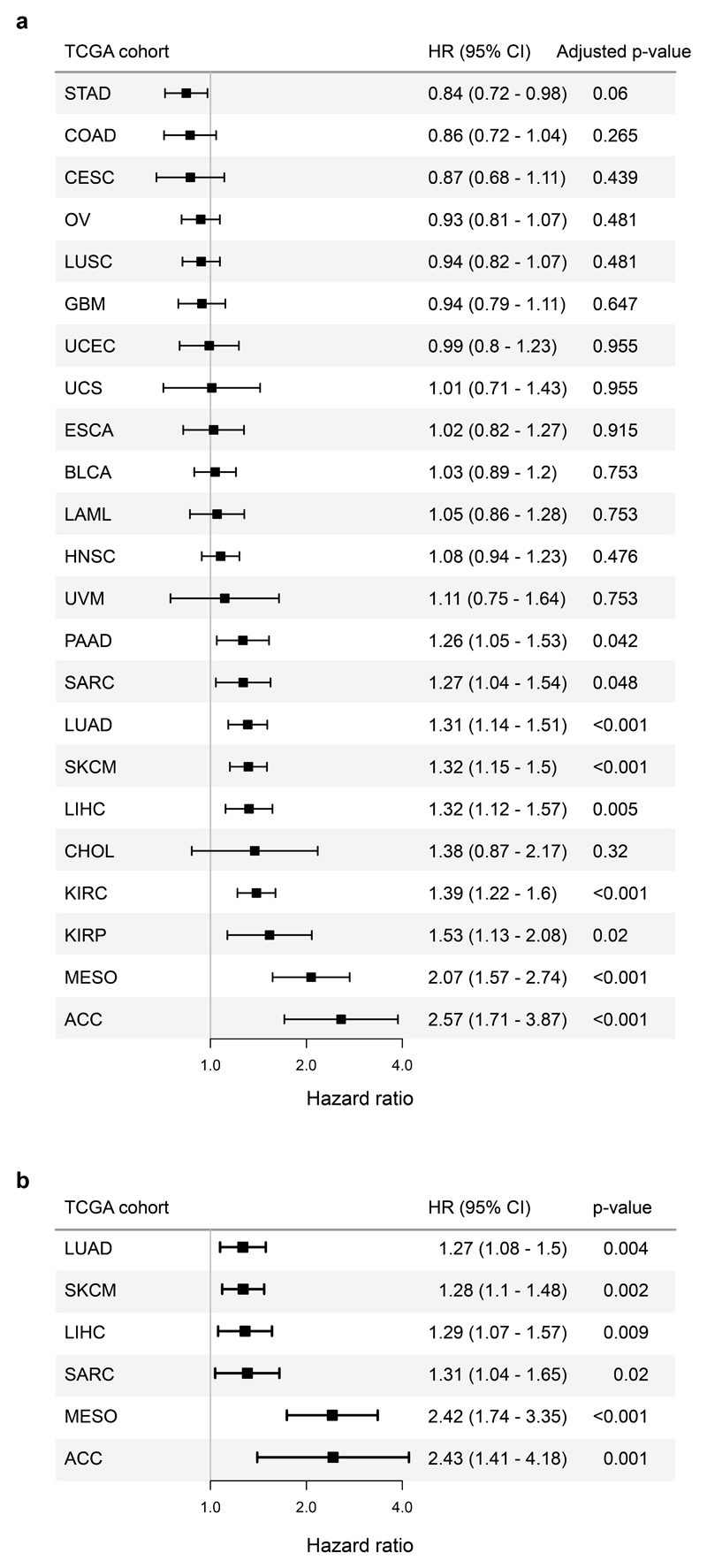
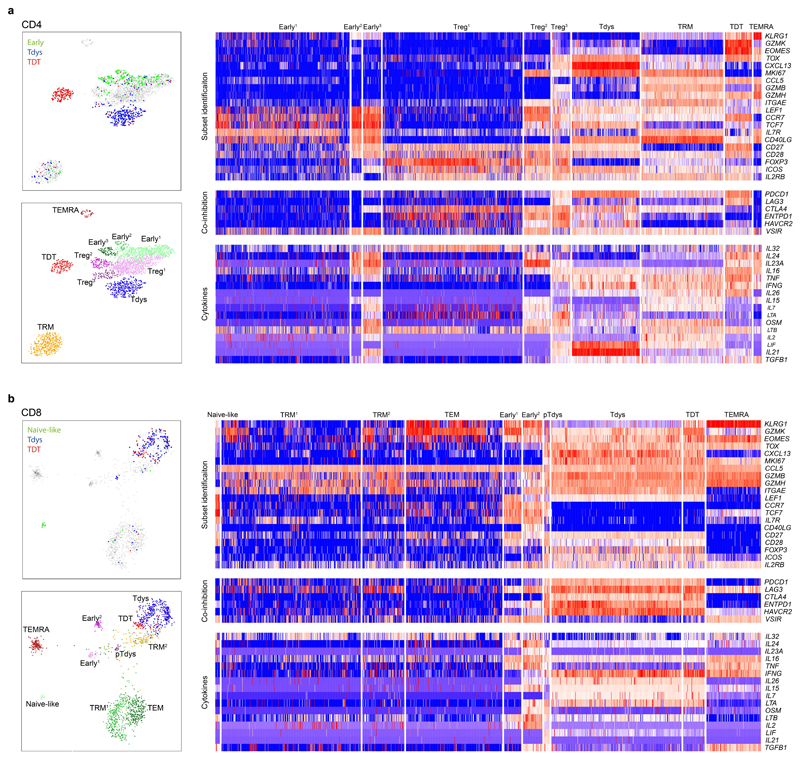
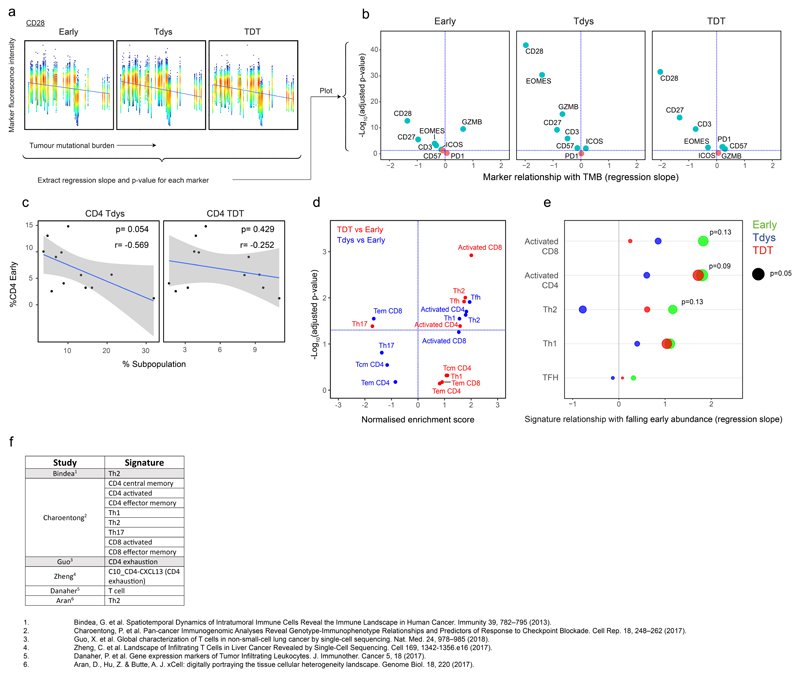
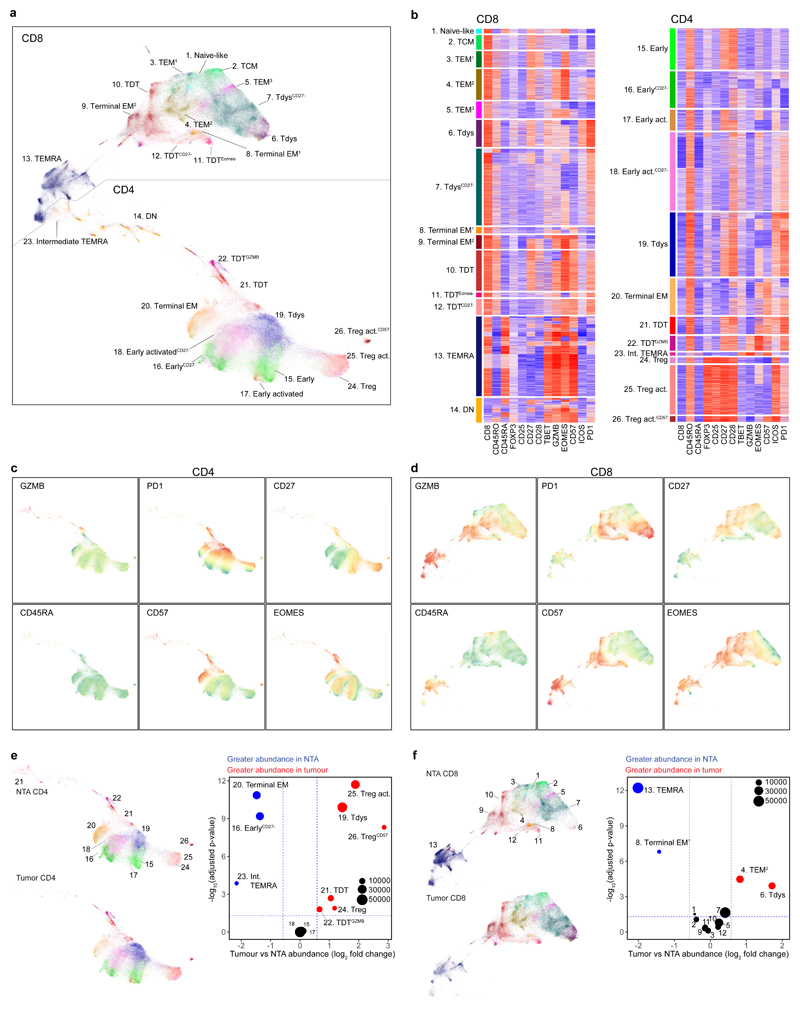
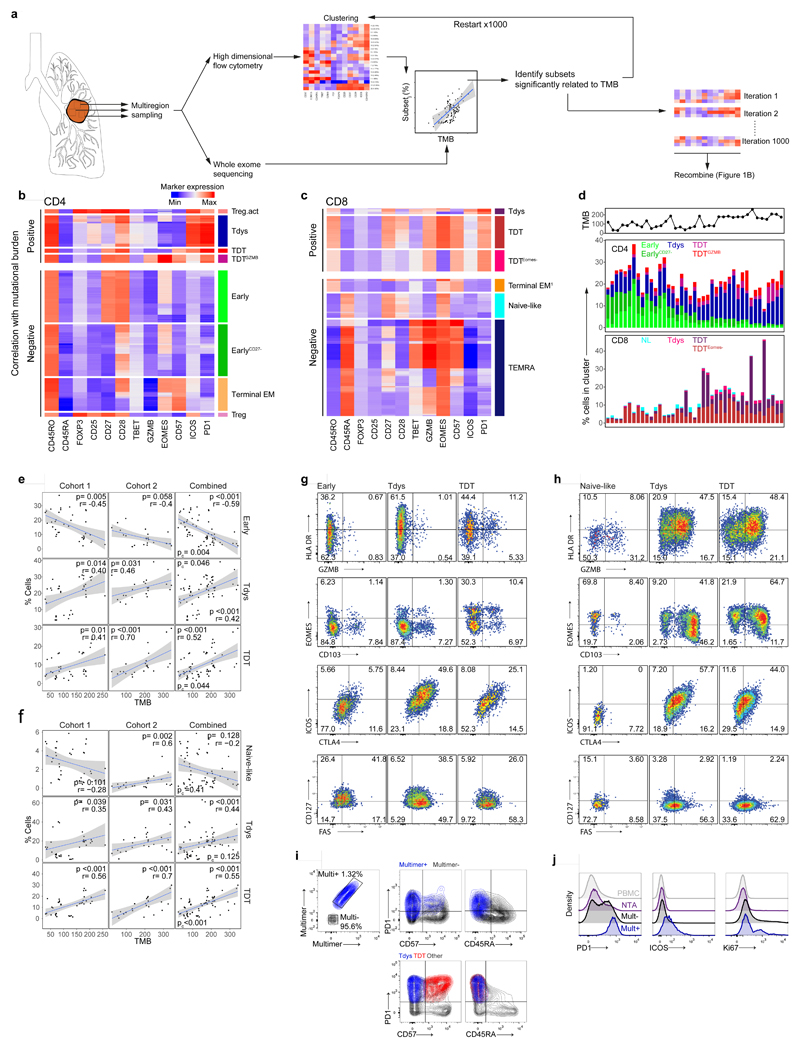
Tumour mutational burden (TMB) predicts immunotherapy outcome in non-small cell lung cancer (NSCLC), consistent with immune recognition of tumour neoantigens. However, persistent antigen exposure is detrimental for T cell function. How TMB affects CD4 and CD8 T cell differentiation in untreated tumours, and whether this affects patient outcomes is unknown. Here we paired high-dimensional flow cytometry, exome, single-cell and bulk RNA sequencing from patients with resected, untreated NSCLC to examine these relationships. TMB was associated with compartment-wide T cell differentiation skewing, characterized by loss of TCF7-expressing progenitor-like CD4 T cells, and an increased abundance of dysfunctional CD8 and CD4 T cell subsets, with significant phenotypic and transcriptional similarity to neoantigen-reactive CD8 T cells. A gene signature of redistribution from progenitor-like to dysfunctional states associated with poor survival in lung and other cancer cohorts. Single-cell characterization of these populations informs potential strategies for therapeutic manipulation in NSCLC.
Conflict of interest statement
Competing interest statement S.A.Q., K.S.P. and C.S. are co-founders of Achilles Therapeutics. C.S. receives grant support from Pfizer, AstraZeneca, BMS, Roche-Ventana and Boehringer-Ingelheim. C.S. has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, BMS, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, and the Sarah Cannon Research Institute. C.S. is a shareholder of Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options in and is co-founder of Achilles Therapeutics. R.R., N.M. and G.A.W. have stock options in and have consulted for Achilles Therapeutics. J.L.R and M.A.B have consulted for Achilles Therapeutics. P.D.B and M.W.S are employees of Achilles Therapeutics.
Figures
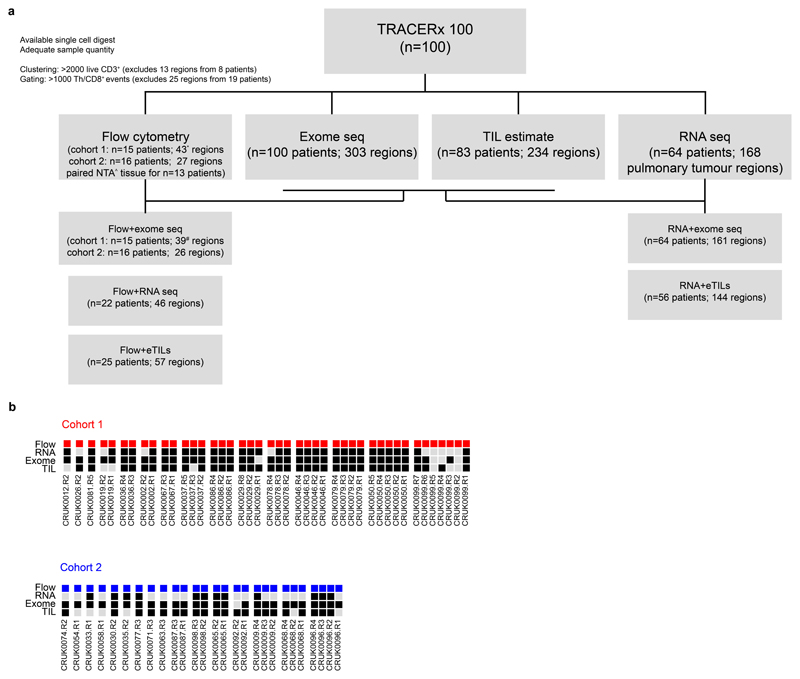
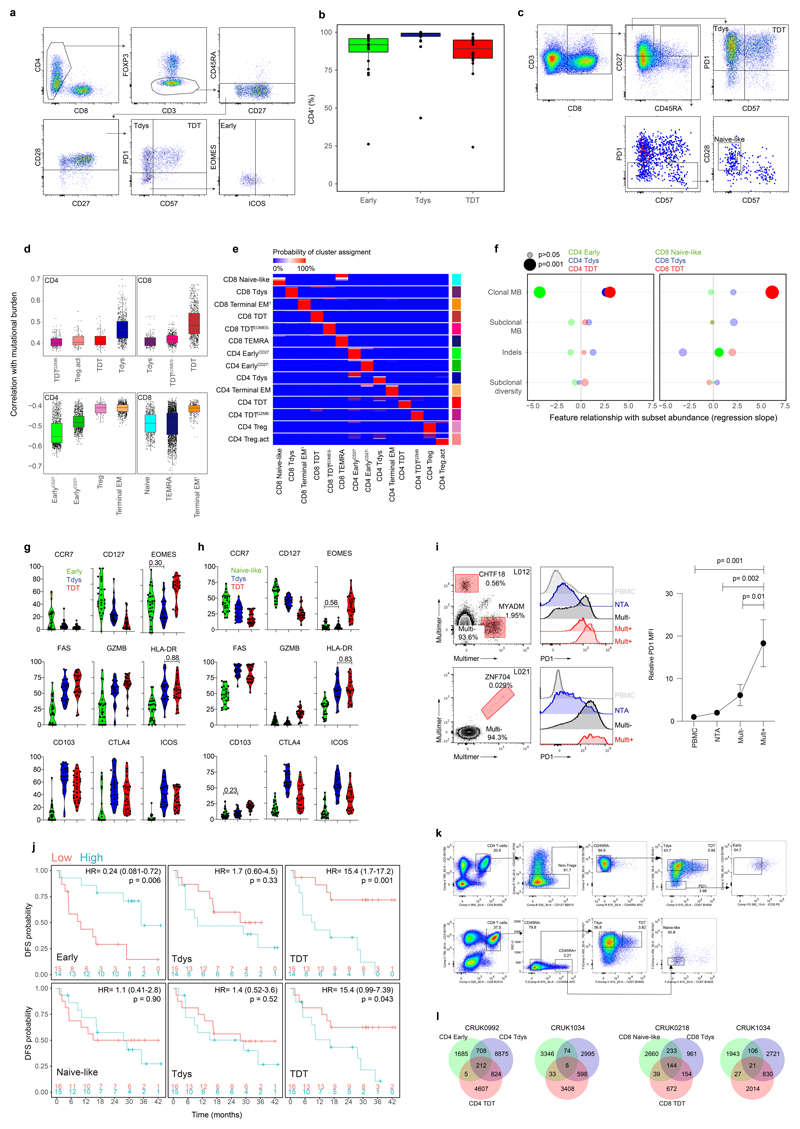
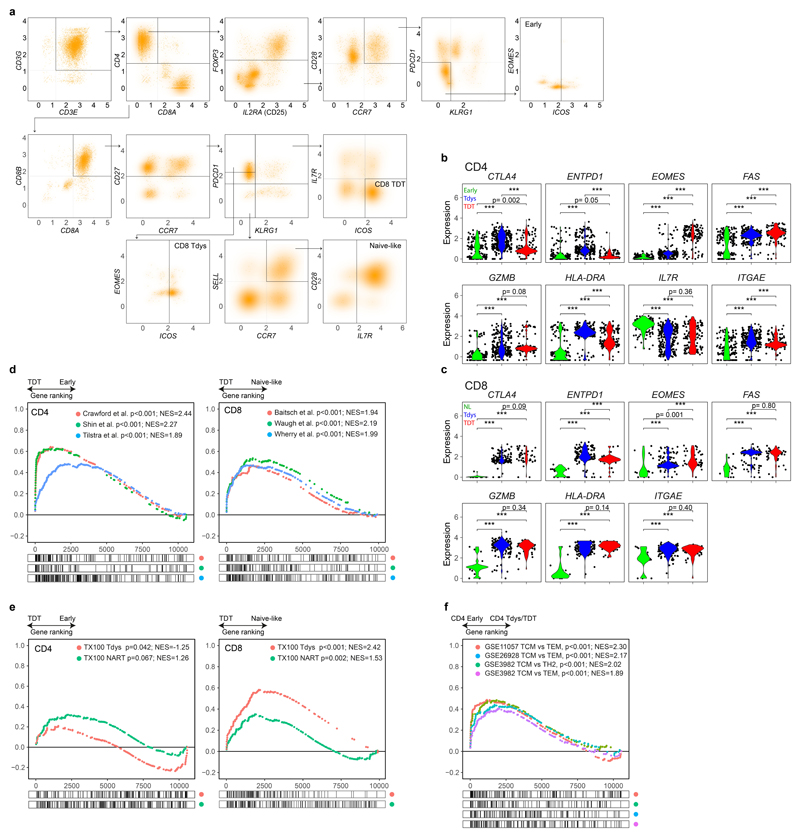
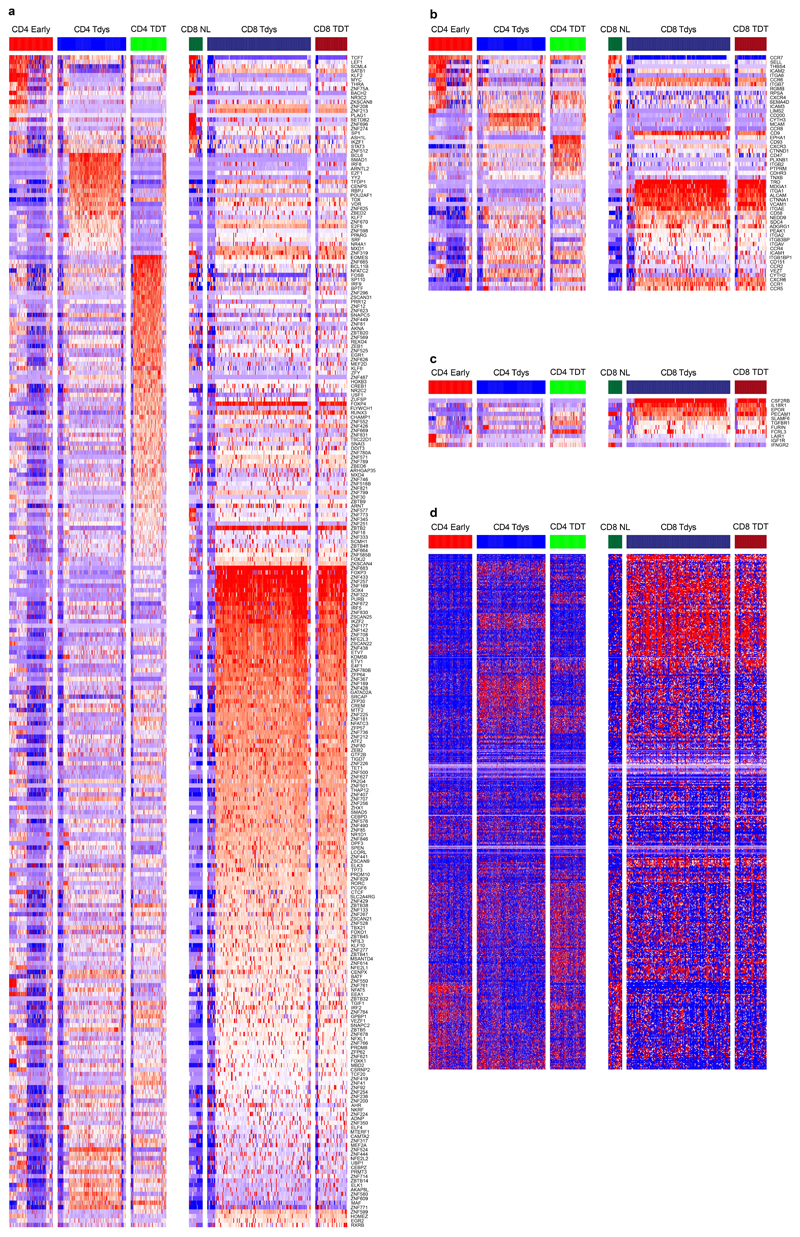
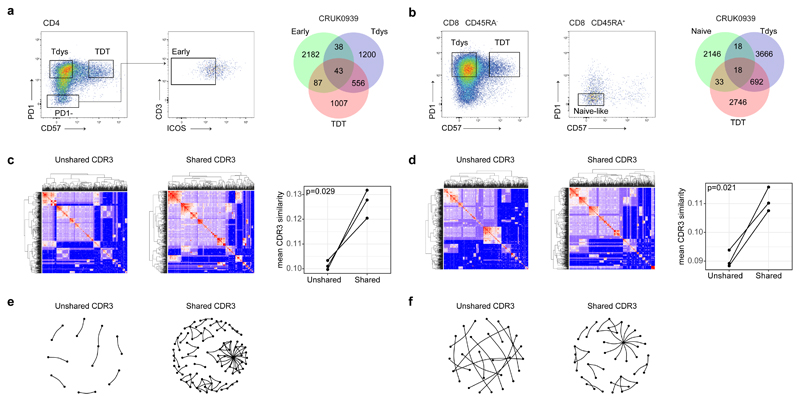
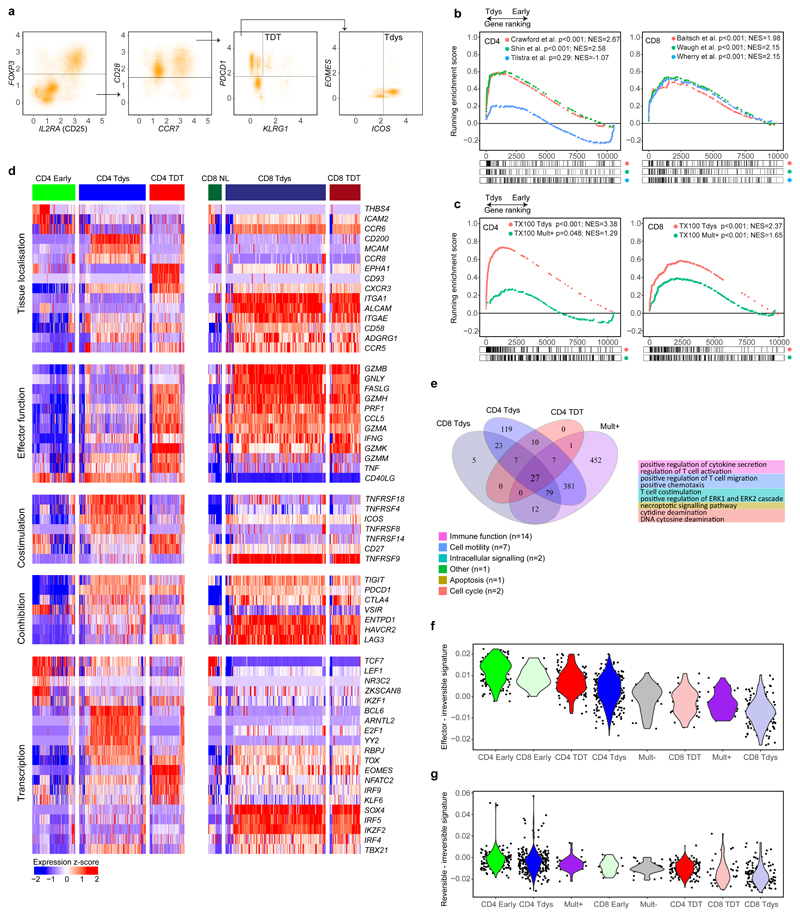
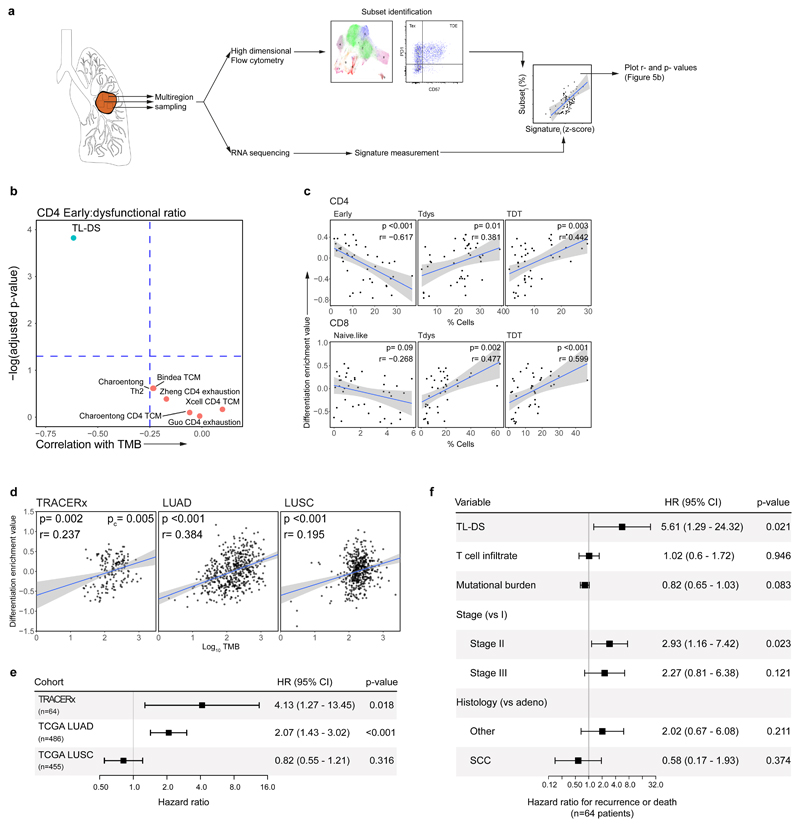
References
-
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (80-) 2015;348:69–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 617844/ERC_/European Research Council/International
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- 20466/CRUK_/Cancer Research UK/United Kingdom
- FC001169/MRC_/Medical Research Council/United Kingdom
- 30194/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- A22246/CRUK_/Cancer Research UK/United Kingdom
- 24314/CRUK_/Cancer Research UK/United Kingdom
- MR/M009033/1/MRC_/Medical Research Council/United Kingdom
- 22246/CRUK_/Cancer Research UK/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- MR/P014712/2/MRC_/Medical Research Council/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- 25223/CRUK_/Cancer Research UK/United Kingdom
- MR/P014712/1/MRC_/Medical Research Council/United Kingdom
- 20764/CRUK_/Cancer Research UK/United Kingdom
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- A20465/CRUK_/Cancer Research UK/United Kingdom
- FC001202/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Research Materials
