

Skin microbiota in health and disease: From sequencing to biology
- PMID: 32804417
- PMCID: PMC7589227
- DOI: 10.1111/1346-8138.15536
Skin microbiota in health and disease: From sequencing to biology
Abstract
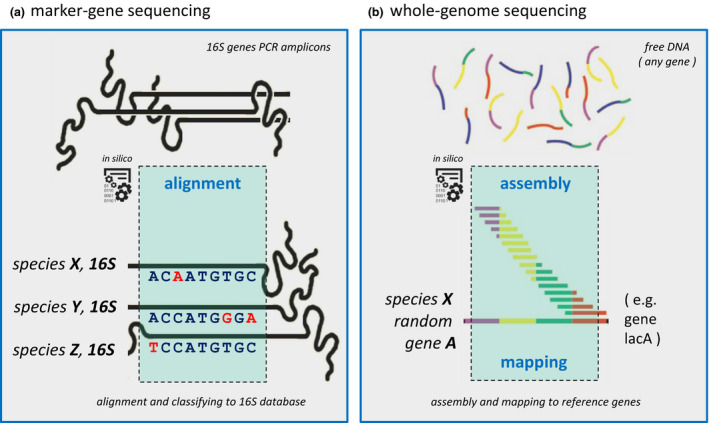
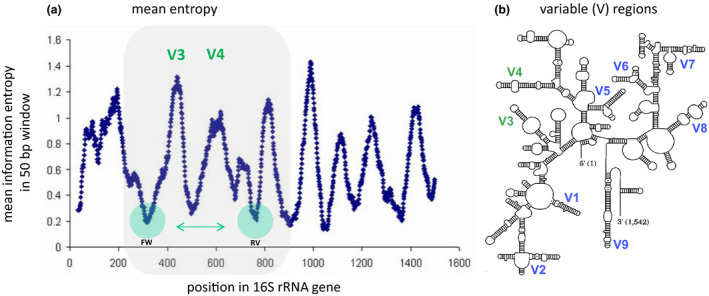
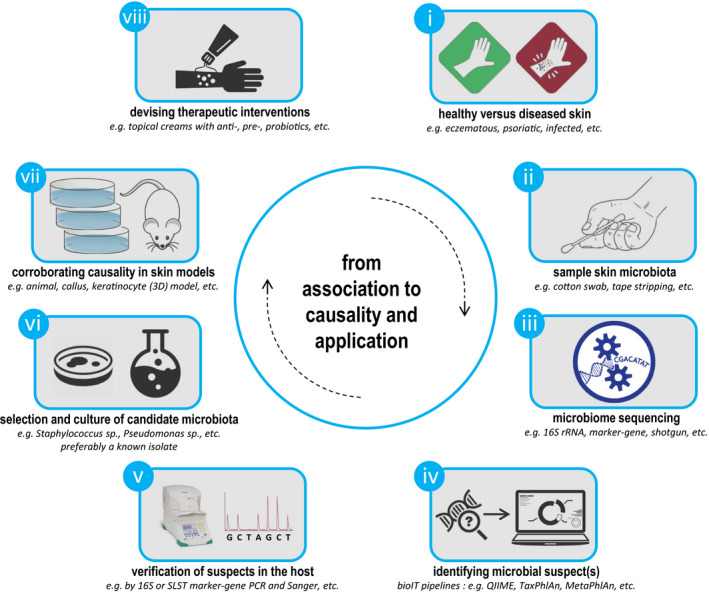
Microbiota live in a closely regulated interaction with their environment, and vice versa. The presence and absence of microbial entities is greatly influenced by features of the niche in which they thrive. Characteristic of this phenomenon is that different human skin sites harbor niche-specific communities of microbes. Microbial diversity is considerable, and the current challenge lies in determining which microbes and (corresponding) functionality are of importance to a given ecological niche. Furthermore, as there is increasing evidence of microbial involvement in health and disease, the need arises to fundamentally understand microbiome processes for application in health care, nutrition and personal care products (e.g. diet, cosmetics, probiotics). This review provides a current overview of state-of-the-art sequencing-based techniques and corresponding data analysis methodology for profiling of complex microbial communities. Furthermore, we also summarize the existing knowledge regarding cutaneous microbiota and their human host for a wide range of skin diseases.
Keywords: bioinformatics; cutaneous diseases; microbiomics; next-generation sequencing; skin microbiota.
© 2020 The Authors. The Journal of Dermatology published by John Wiley & Sons Australia, Ltd on behalf of Japanese Dermatological Association.
Conflict of interest statement
None declared.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
