

Mitochondrial energetic impairment in a patient with late-onset glutaric acidemia Type 2
- PMID: 32804429
- PMCID: PMC8543298
- DOI: 10.1002/ajmg.a.61786
Mitochondrial energetic impairment in a patient with late-onset glutaric acidemia Type 2
Abstract
Glutaric acidemia type 2 (GA2), also called multiple acyl-CoA dehydrogenase deficiency, is an autosomal recessive disorder of fatty acid, amino acid, and choline metabolism resulting in excretion of multiple organic acids and glycine conjugates as well as elevation of various plasma acylcarnitine species (C4-C18). It is caused by mutations in the ETFA, ETFB, or ETFDH genes which are involved in the transfer of electrons from 11 flavin-containing dehydrogenases to Coenzyme Q10 (CoQ10 ) of the mitochondrial electron transport chain (ETC). We report a patient who was originally reported as the first case with primary myopathic CoQ10 deficiency when he presented at 11.5 years with exercise intolerance and myopathy that improved after treatment with ubiquinone and carnitine. At age 23, his symptoms relapsed despite increasing doses of ubiquinone and he was shown to have biallelic mutations in the ETFDH gene. Treatment with riboflavin was started and ubiquinone was changed to ubiquinol. After 4 months, the patient recovered his muscle strength with normalization of laboratory exams and exercise tolerance. Functional studies on fibroblasts revealed decreased levels of ETFDH as well as of very long-chain acyl-CoA dehydrogenase and trifunctional protein α. In addition, the mitochondrial mass was decreased, with increased formation of reactive oxygen species and oxygen consumption rate, but with a decreased spared respiratory capacity, and decreased adenosine triphosphate level. These findings of widespread dysfunction of fatty acid oxidation and ETC enzymes support the impairment of a larger mitochondrial ETC supercomplex in our patient.
Keywords: ETFDH; electron transport chain; glutaric acidemia type 2; mitochondria.
© 2020 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST
The authors of this manuscript have no conflicts of interest to declare.
Figures
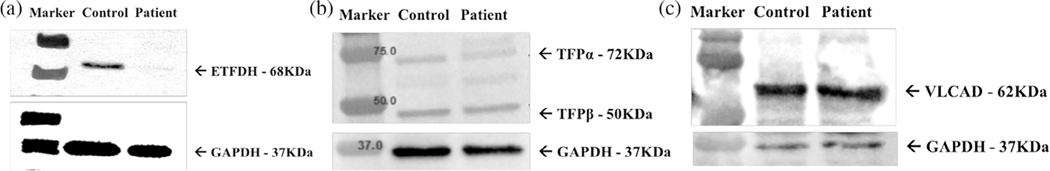
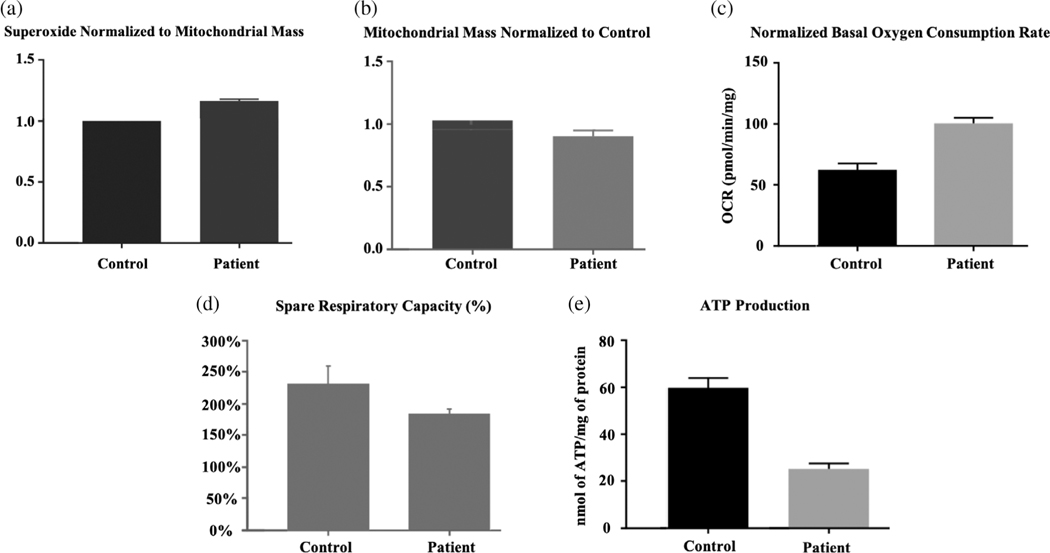
References
-
- Amendt BA, & Rhead WJ (1986). The multiple acyl-coenzyme A dehydrogenation disorders, glutaric aciduria type II and ethylmalonic-adipic aciduria. Mitochondrial fatty acid oxidation, acyl-coenzyme A dehydrogenase, and electron transfer flavoprotein activities in fibroblasts. The Journal of Clinical Investigation, 78(1), 205–213. 10.1172/JCI112553 - DOI - PMC - PubMed
-
- Cornelius N, Byron C, Hargreaves I, Fernandez Guerra P, Furdek AK, Land J, ... Olsen RKJ (2013). Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple acyl-CoA dehydrogenation deficiency. Human Molecular Genetics, 22(19), 3819–3827. 10.1093/hmg/ddt232 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
