

Deconvolving the contributions of cell-type heterogeneity on cortical gene expression
- PMID: 32804935
- PMCID: PMC7451979
- DOI: 10.1371/journal.pcbi.1008120
Deconvolving the contributions of cell-type heterogeneity on cortical gene expression
Abstract
Complexity of cell-type composition has created much skepticism surrounding the interpretation of bulk tissue transcriptomic studies. Recent studies have shown that deconvolution algorithms can be applied to computationally estimate cell-type proportions from gene expression data of bulk blood samples, but their performance when applied to brain tissue is unclear. Here, we have generated an immunohistochemistry (IHC) dataset for five major cell-types from brain tissue of 70 individuals, who also have bulk cortical gene expression data. With the IHC data as the benchmark, this resource enables quantitative assessment of deconvolution algorithms for brain tissue. We apply existing deconvolution algorithms to brain tissue by using marker sets derived from human brain single cell and cell-sorted RNA-seq data. We show that these algorithms can indeed produce informative estimates of constituent cell-type proportions. In fact, neuronal subpopulations can also be estimated from bulk brain tissue samples. Further, we show that including the cell-type proportion estimates as confounding factors is important for reducing false associations between Alzheimer's disease phenotypes and gene expression. Lastly, we demonstrate that using more accurate marker sets can substantially improve statistical power in detecting cell-type specific expression quantitative trait loci (eQTLs).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
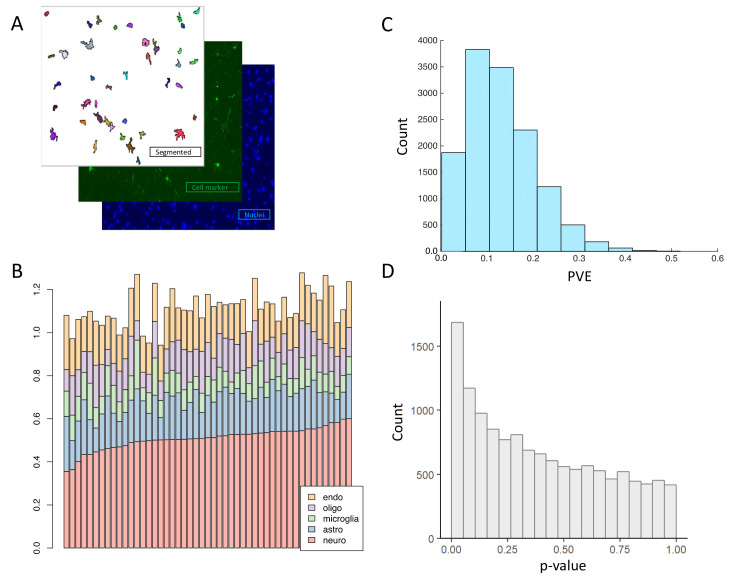


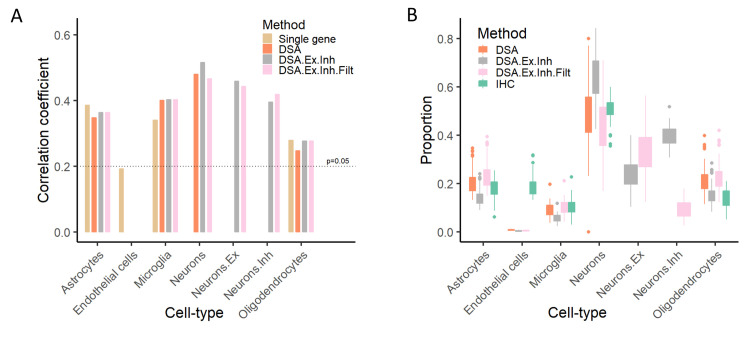

References
-
- Hunt GJ, Freytag S, Bahlo M, Gagnon-Bartsch JA. dtangle: accurate and fast cell-type deconvolution. bioRxiv. 2018. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
