

Systematic identification of functional SNPs interrupting 3'UTR polyadenylation signals
- PMID: 32804959
- PMCID: PMC7451987
- DOI: 10.1371/journal.pgen.1008977
Systematic identification of functional SNPs interrupting 3'UTR polyadenylation signals
Abstract
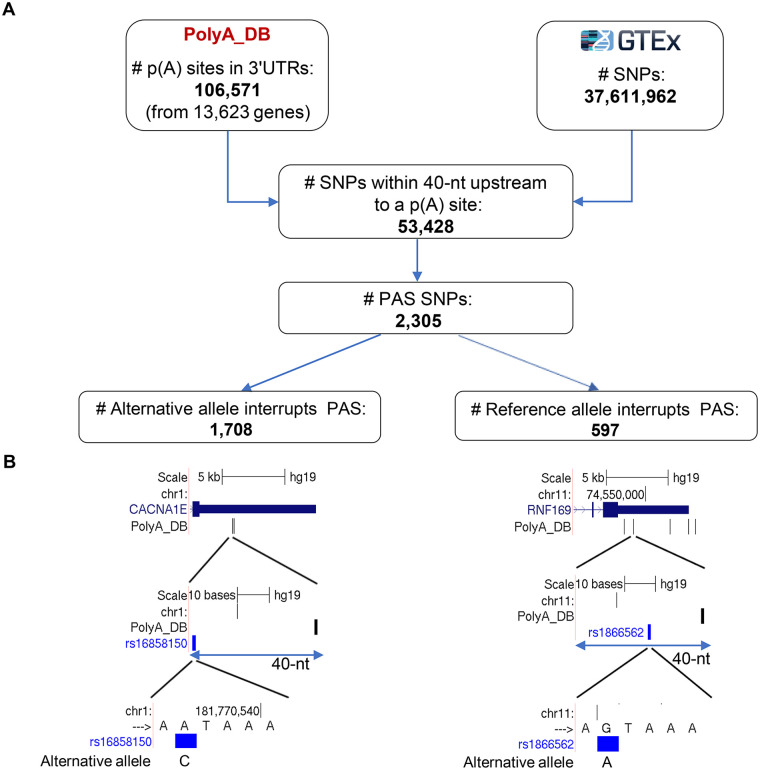
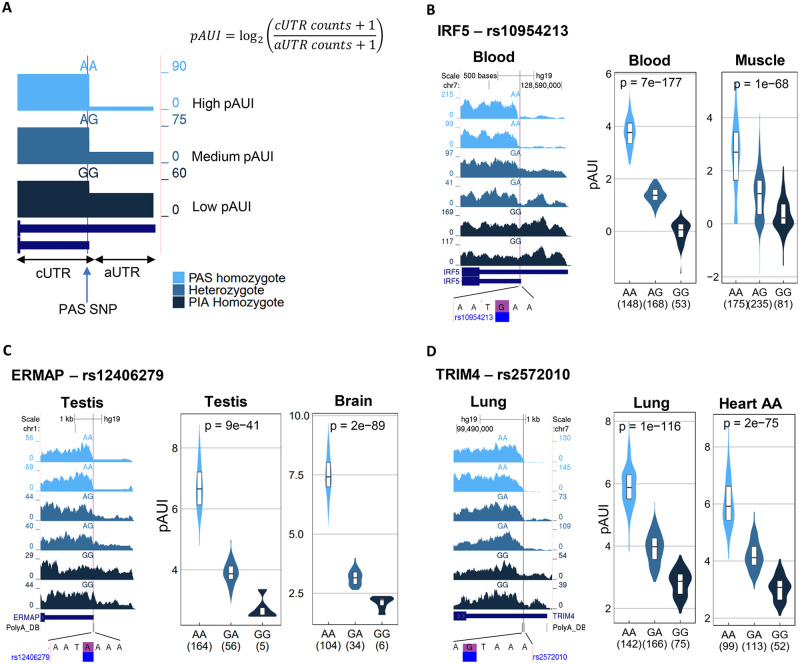
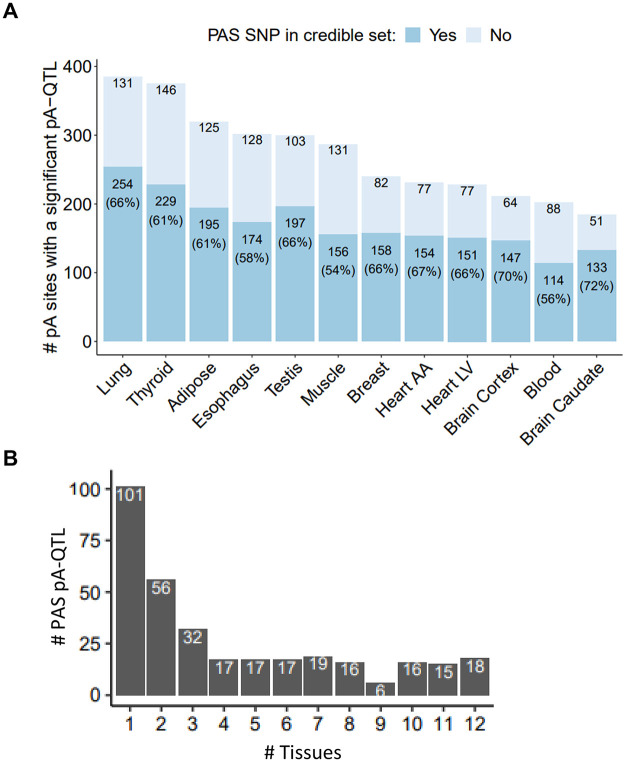
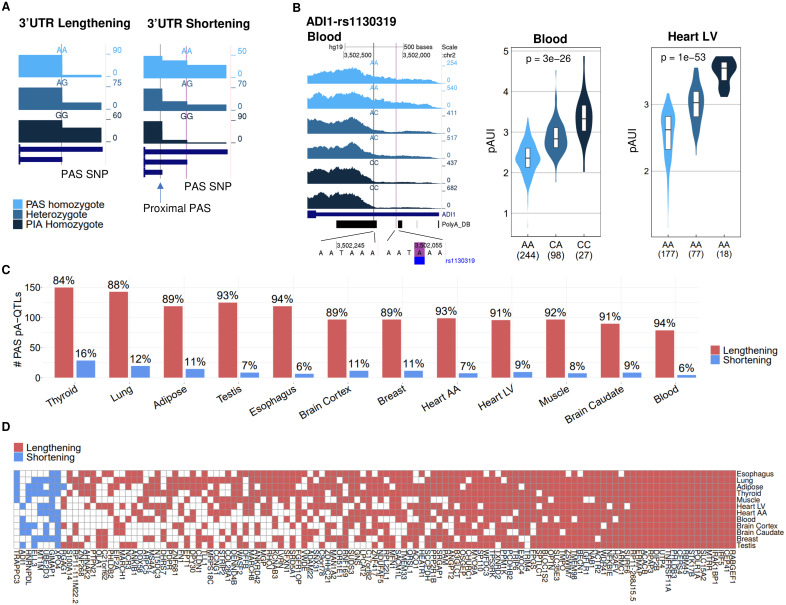
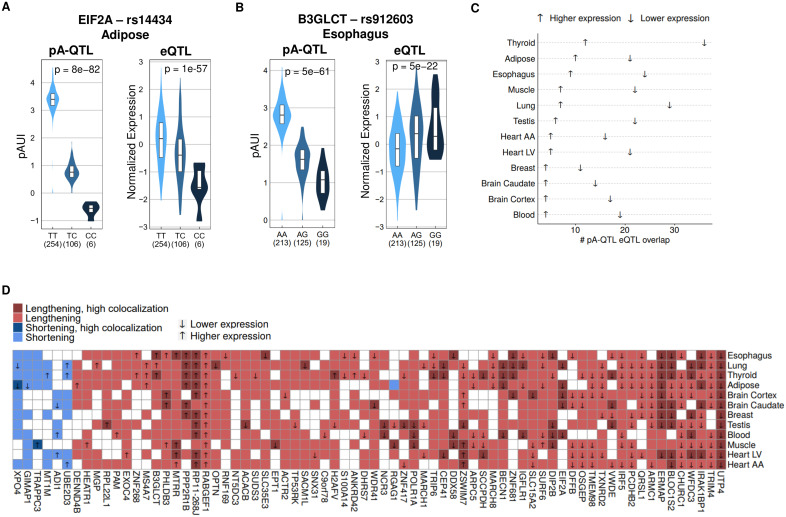
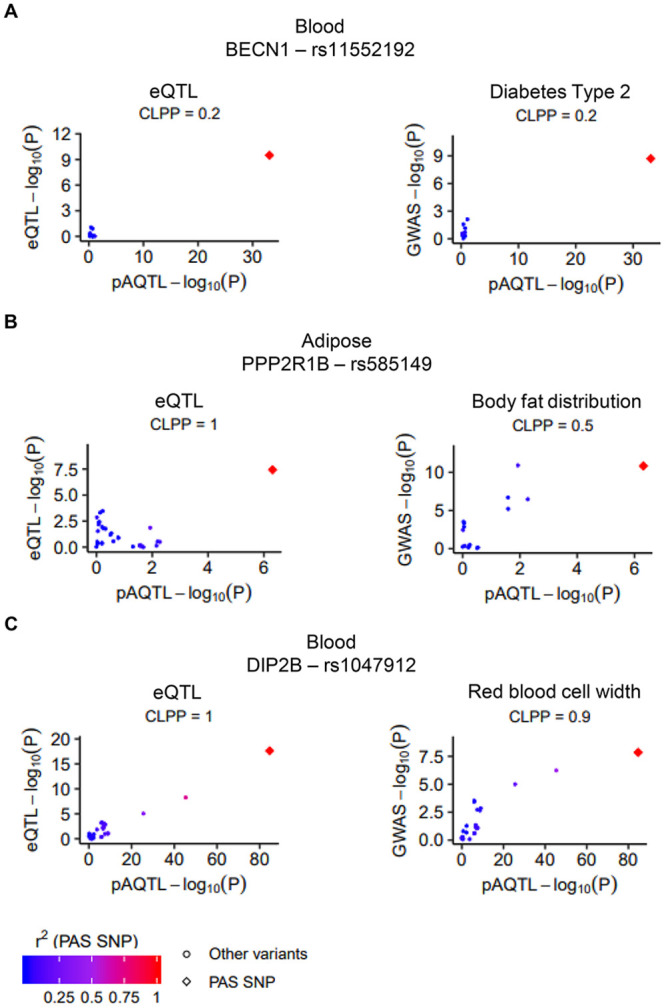
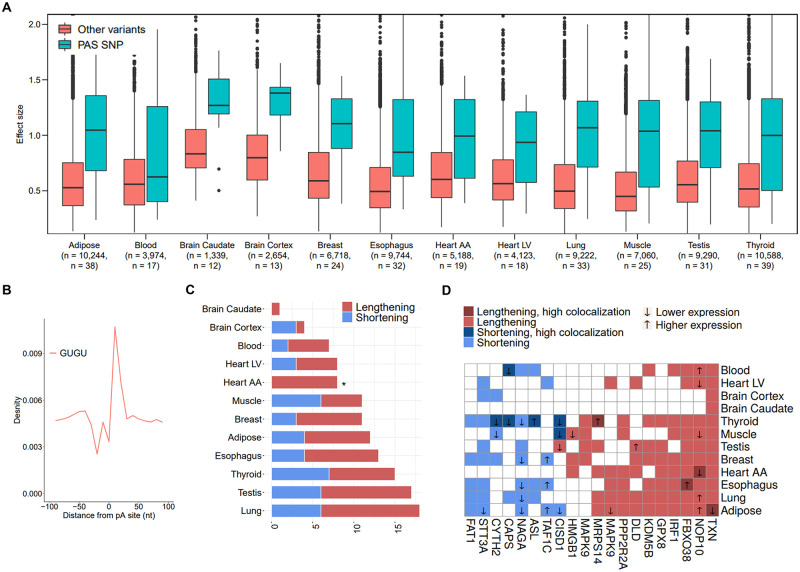
Alternative polyadenylation (APA) is emerging as a widespread regulatory layer since the majority of human protein-coding genes contain several polyadenylation (p(A)) sites in their 3'UTRs. By generating isoforms with different 3'UTR length, APA potentially affects mRNA stability, translation efficiency, nuclear export, and cellular localization. Polyadenylation sites are regulated by adjacent RNA cis-regulatory elements, the principals among them are the polyadenylation signal (PAS) AAUAAA and its main variant AUUAAA, typically located ~20-nt upstream of the p(A) site. Mutations in PAS and other auxiliary poly(A) cis-elements in the 3'UTR of several genes have been shown to cause human Mendelian diseases, and to date, only a few common SNPs that regulate APA were associated with complex diseases. Here, we systematically searched for SNPs that affect gene expression and human traits by modulation of 3'UTR APA. First, focusing on the variants most likely to exert the strongest effect, we identified 2,305 SNPs that interrupt the canonical PAS or its main variant. Implementing pA-QTL tests using GTEx RNA-seq data, we identified 330 PAS SNPs (called PAS pA-QTLs) that were significantly associated with the usage of their p(A) site. As expected, PAS-interrupting alleles were mostly linked with decreased cleavage at their p(A) site and the consequential 3'UTR lengthening. However, interestingly, in ~10% of the cases, the PAS-interrupting allele was associated with increased usage of an upstream p(A) site and 3'UTR shortening. As an indication of the functional effects of these PAS pA-QTLs on gene expression and complex human traits, we observed for few dozens of them marked colocalization with eQTL and/or GWAS signals. The PAS-interrupting alleles linked with 3'UTR lengthening were also strongly associated with decreased gene expression, indicating that shorter isoforms generated by APA are generally more stable than longer ones. Last, we carried out an extended, genome-wide analysis of 3'UTR variants and detected thousands of additional pA-QTLs having weaker effects compared to the PAS pA-QTLs.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
3'aQTL-atlas: an atlas of 3'UTR alternative polyadenylation quantitative trait loci across human normal tissues.Nucleic Acids Res. 2022 Jan 7;50(D1):D39-D45. doi: 10.1093/nar/gkab740. Nucleic Acids Res. 2022. PMID: 34432052 Free PMC article.
-
Alternative cleavage and polyadenylation in spermatogenesis connects chromatin regulation with post-transcriptional control.BMC Biol. 2016 Jan 22;14:6. doi: 10.1186/s12915-016-0229-6. BMC Biol. 2016. PMID: 26801249 Free PMC article.
-
Complex Selection on Human Polyadenylation Signals Revealed by Polymorphism and Divergence Data.Genome Biol Evol. 2016 Jul 2;8(6):1971-9. doi: 10.1093/gbe/evw137. Genome Biol Evol. 2016. PMID: 27324920 Free PMC article.
-
Implications of polyadenylation in health and disease.Nucleus. 2014;5(6):508-19. doi: 10.4161/nucl.36360. Epub 2014 Oct 31. Nucleus. 2014. PMID: 25484187 Free PMC article. Review.
-
Regulation and function of alternative polyadenylation in development and differentiation.RNA Biol. 2023 Jan;20(1):908-925. doi: 10.1080/15476286.2023.2275109. Epub 2023 Oct 31. RNA Biol. 2023. PMID: 37906624 Free PMC article. Review.
Cited by
-
CRISPRpas: programmable regulation of alternative polyadenylation by dCas9.Nucleic Acids Res. 2022 Mar 21;50(5):e25. doi: 10.1093/nar/gkab519. Nucleic Acids Res. 2022. PMID: 34244761 Free PMC article.
-
Analysis Polyadenylation Signal Usage in Sus scrofa.Animals (Basel). 2022 Jan 13;12(2):194. doi: 10.3390/ani12020194. Animals (Basel). 2022. PMID: 35049816 Free PMC article.
-
The Advances in Deep Learning Modeling of Polyadenylation Codes.Wiley Interdiscip Rev RNA. 2025 May-Jun;16(3):e70017. doi: 10.1002/wrna.70017. Wiley Interdiscip Rev RNA. 2025. PMID: 40468587 Free PMC article. Review.
-
Deep learning of human polyadenylation sites at nucleotide resolution reveals molecular determinants of site usage and relevance in disease.Nat Commun. 2023 Nov 15;14(1):7378. doi: 10.1038/s41467-023-43266-3. Nat Commun. 2023. PMID: 37968271 Free PMC article.
-
Glucocorticoids mediate transcriptome-wide alternative polyadenylation: Potential mechanistic and clinical implications.Clin Transl Sci. 2022 Nov;15(11):2758-2771. doi: 10.1111/cts.13402. Epub 2022 Sep 20. Clin Transl Sci. 2022. PMID: 36128656 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
