

Two mouse models carrying truncating mutations in Magel2 show distinct phenotypes
- PMID: 32804975
- PMCID: PMC7430741
- DOI: 10.1371/journal.pone.0237814
Two mouse models carrying truncating mutations in Magel2 show distinct phenotypes
Abstract
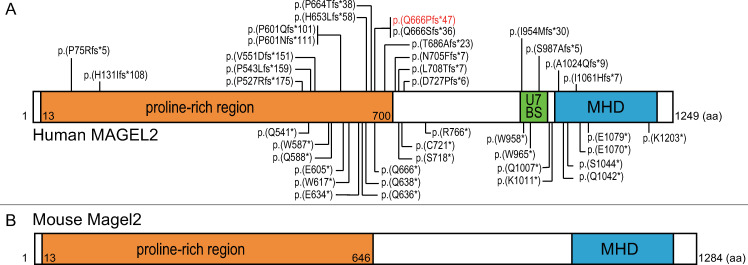
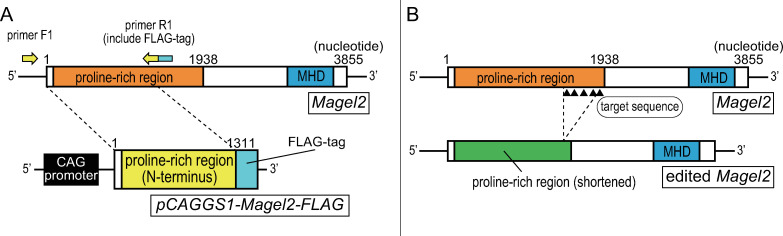
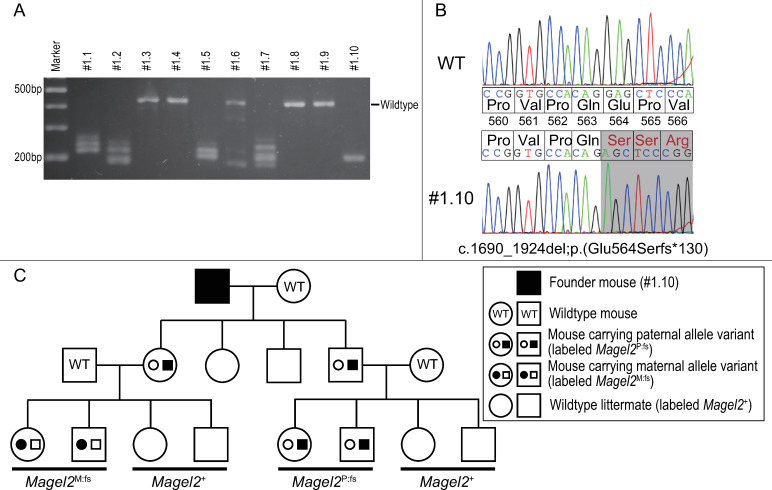
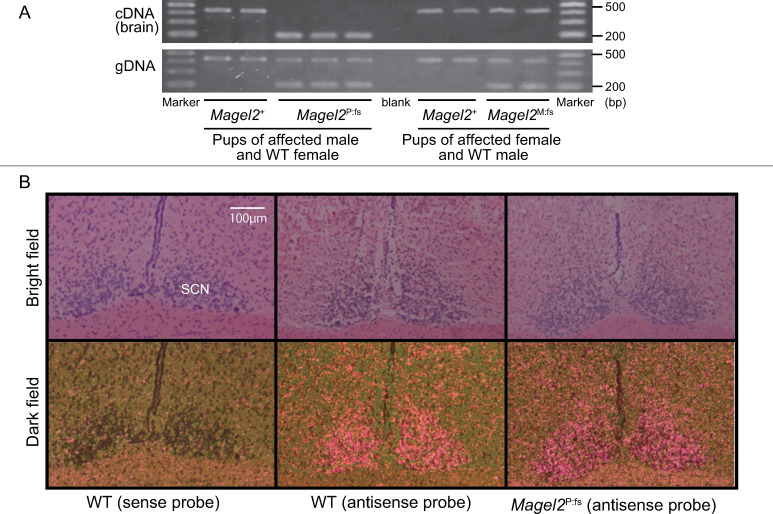
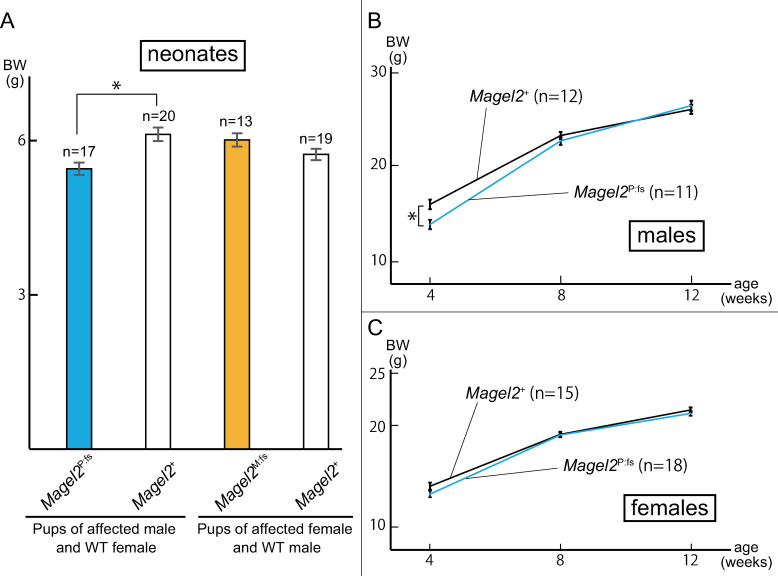
Schaaf-Yang syndrome (SYS) is a neurodevelopmental disorder caused by truncating variants in the paternal allele of MAGEL2, located in the Prader-Willi critical region, 15q11-q13. Although the phenotypes of SYS overlap those of Prader-Willi syndrome (PWS), including neonatal hypotonia, feeding problems, and developmental delay/intellectual disability, SYS patients show autism spectrum disorder and joint contractures, which are atypical phenotypes for PWS. Therefore, we hypothesized that the truncated Magel2 protein could potentially produce gain-of-function toxic effects. To test the hypothesis, we generated two engineered mouse models; one, an overexpression model that expressed the N-terminal region of Magel2 that was FLAG tagged with a strong ubiquitous promoter, and another, a genome-edited model that carried a truncating variant in Magel2 generated using the CRISPR/Cas9 system. In the overexpression model, all transgenic mice died in the fetal or neonatal period indicating embryonic or neonatal lethality of the transgene. Therefore, overexpression of the truncated Magel2 could show toxic effects. In the genome-edited model, we generated a mouse model carrying a frameshift variant (c.1690_1924del; p(Glu564Serfs*130)) in Magel2. Model mice carrying the frameshift variant in the paternal or maternal allele of Magel2 were termed Magel2P:fs and Magel2M:fs, respectively. The imprinted expression and spatial distribution of truncating Magel2 transcripts in the brain were maintained. Although neonatal Magel2P:fs mice were lighter than wildtype littermates, Magel2P:fs males and females weighed the same as their wildtype littermates by eight and four weeks of age, respectively. Collectively, the overexpression mouse model may recapitulate fetal or neonatal death, which are the severest phenotypes for SYS. In contrast, the genome-edited mouse model maintains genomic imprinting and distribution of truncated Magel2 transcripts in the brain, but only partially recapitulates SYS phenotypes. Therefore, our results imply that simple gain-of-function toxic effects may not explain the patho-mechanism of SYS, but rather suggest a range of effects due to Magel2 variants as in human SYS patients.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
