

Inferring cancer progression from Single-Cell Sequencing while allowing mutation losses
- PMID: 32805010
- PMCID: PMC8058767
- DOI: 10.1093/bioinformatics/btaa722
Inferring cancer progression from Single-Cell Sequencing while allowing mutation losses
Abstract
Motivation: In recent years, the well-known Infinite Sites Assumption has been a fundamental feature of computational methods devised for reconstructing tumor phylogenies and inferring cancer progressions. However, recent studies leveraging single-cell sequencing (SCS) techniques have shown evidence of the widespread recurrence and, especially, loss of mutations in several tumor samples. While there exist established computational methods that infer phylogenies with mutation losses, there remain some advancements to be made.
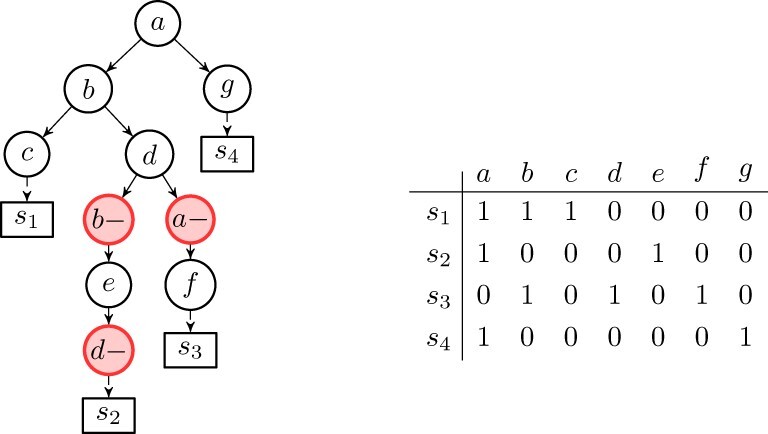
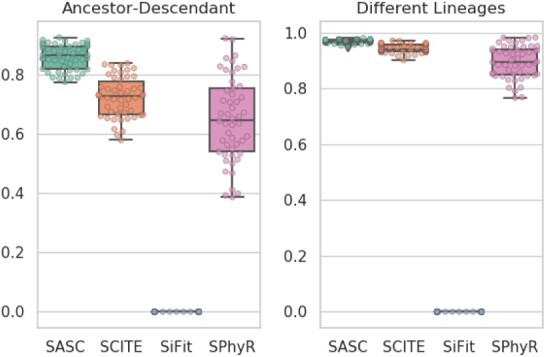
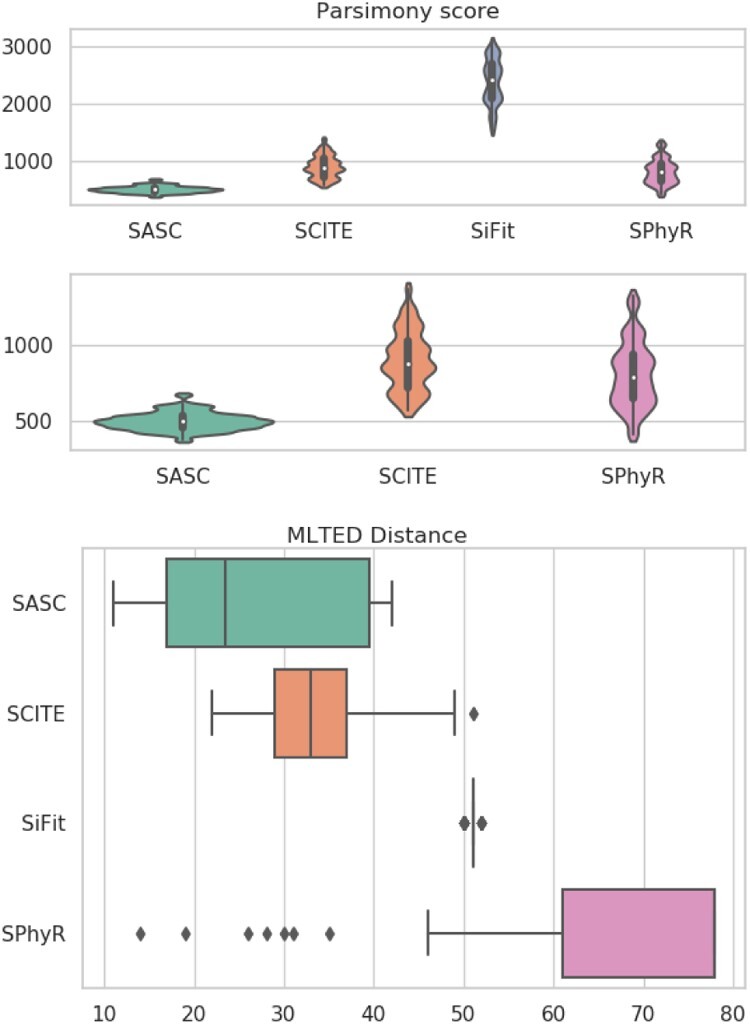
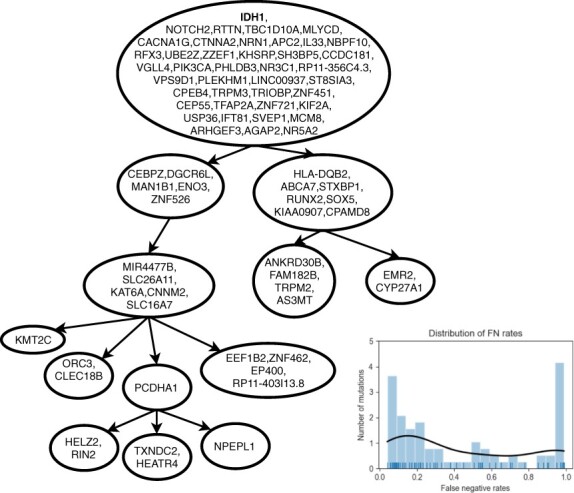
Results: We present Simulated Annealing Single-Cell inference (SASC): a new and robust approach based on simulated annealing for the inference of cancer progression from SCS datasets. In particular, we introduce an extension of the model of evolution where mutations are only accumulated, by allowing also a limited amount of mutation loss in the evolutionary history of the tumor: the Dollo-k model. We demonstrate that SASC achieves high levels of accuracy when tested on both simulated and real datasets and in comparison with some other available methods.
Availability and implementation: The SASC tool is open source and available at https://github.com/sciccolella/sasc.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press.
Figures

References
-
- Bonizzoni P. et al. (2012) The binary perfect phylogeny with persistent characters. Theor. Comput. Sci., 454, 51–63.
-
- Bonizzoni P. et al. (2017) A colored graph approach to perfect phylogeny with persistent characters. Theor. Comput. Sci., 658, 60–73.
-
- Bonizzoni P. et al. (2018) Does relaxing the infinite sites assumption give better tumor phylogenies? An ILP-based comparative approach. IEEE/ACM Trans. Comput. Biol. Bioinform., 16, 1410–1423. - PubMed
-
- Della Vedova G. et al. (2017) Character-based Phylogeny Construction and Its Application to Tumor Evolution. Lecture Notes in Computer Science. Unveiling Dynamics and Complexity - 13th Conference on Computability in Europe, CiE 2017, Turku, Finland, June 12-16, 2017, Proceedings. Vol. 10307, pp. 3–13.
