

Methods for copy number aberration detection from single-cell DNA-sequencing data
- PMID: 32807205
- PMCID: PMC7433197
- DOI: 10.1186/s13059-020-02119-8
Methods for copy number aberration detection from single-cell DNA-sequencing data
Abstract
Copy number aberrations (CNAs), which are pathogenic copy number variations (CNVs), play an important role in the initiation and progression of cancer. Single-cell DNA-sequencing (scDNAseq) technologies produce data that is ideal for inferring CNAs. In this review, we review eight methods that have been developed for detecting CNAs in scDNAseq data, and categorize them according to the steps of a seven-step pipeline that they employ. Furthermore, we review models and methods for evolutionary analyses of CNAs from scDNAseq data and highlight advances and future research directions for computational methods for CNA detection from scDNAseq data.
Keywords: Copy number aberrations; Intra-tumor heterogeneity; Single-cell DNA sequencing; Tumor evolution.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
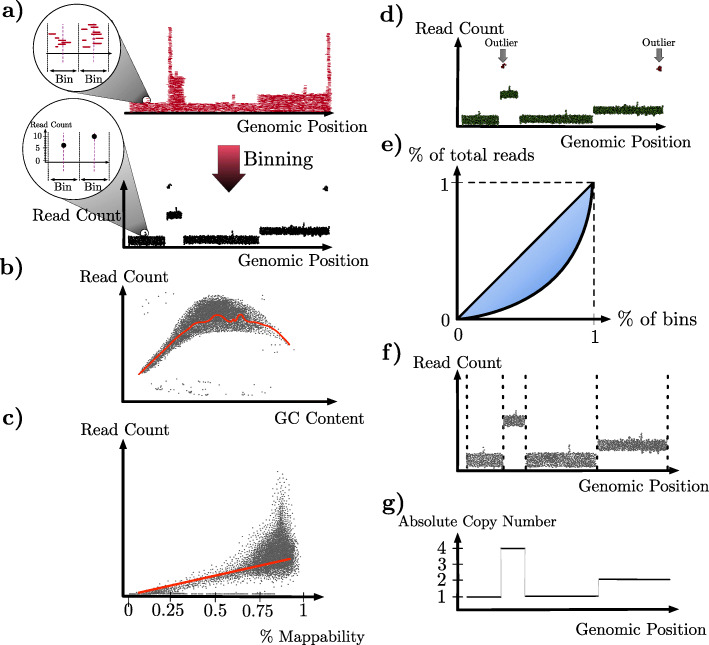
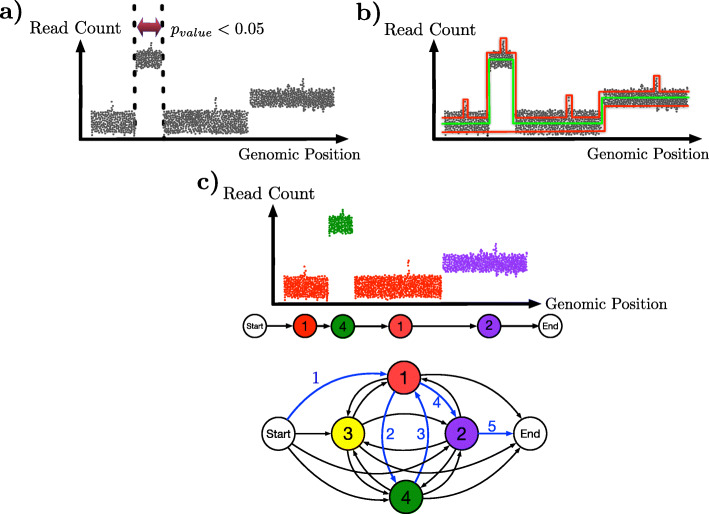
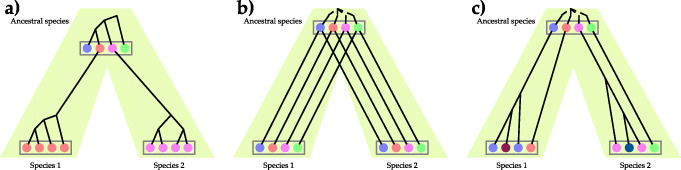
References
-
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–45. - PubMed
-
- Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20(7):404–16. - PubMed
-
- Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Trans Med. 2012;4(127):127–1012710. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
