

Widespread protein lysine acetylation in gut microbiome and its alterations in patients with Crohn's disease
- PMID: 32807798
- PMCID: PMC7431864
- DOI: 10.1038/s41467-020-17916-9
Widespread protein lysine acetylation in gut microbiome and its alterations in patients with Crohn's disease
Abstract
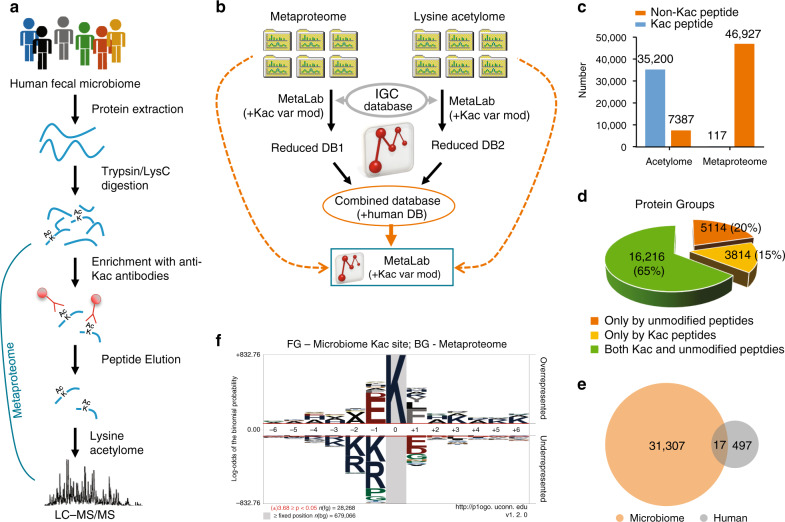
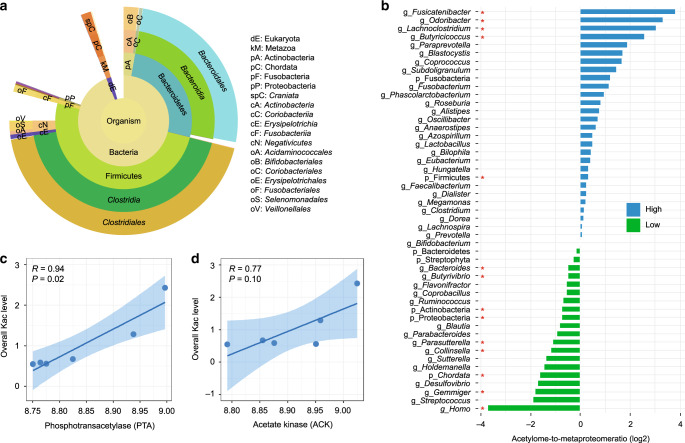
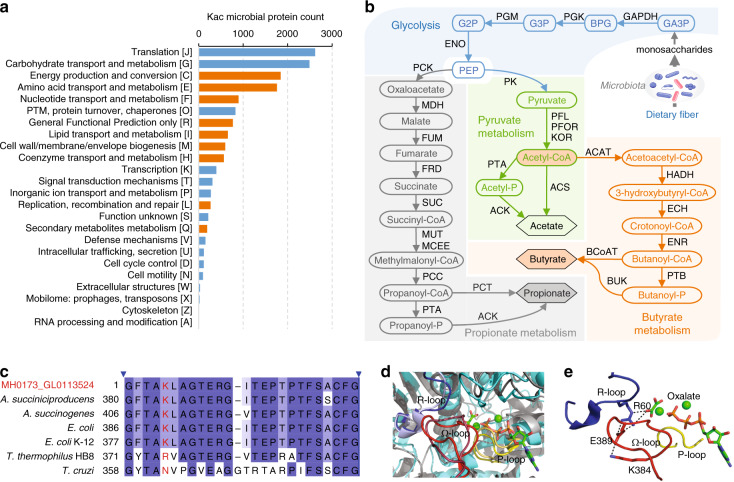
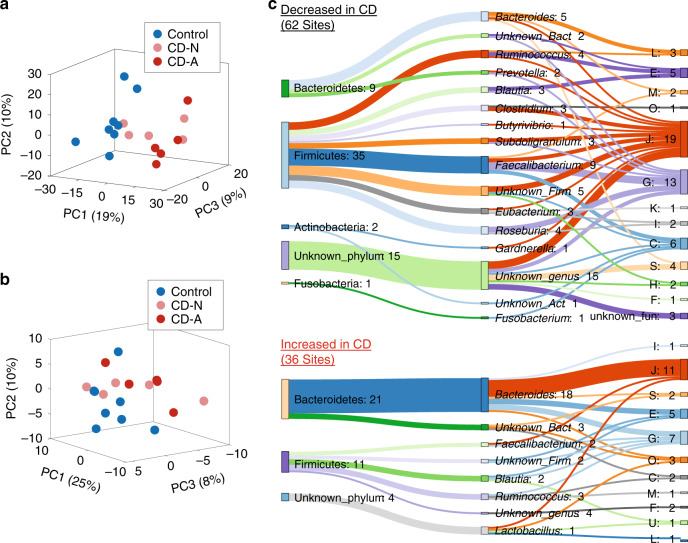
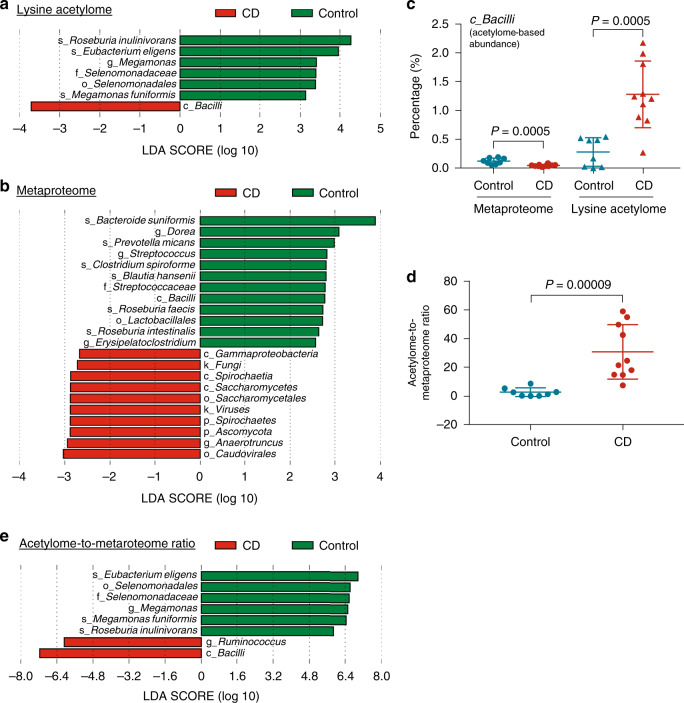
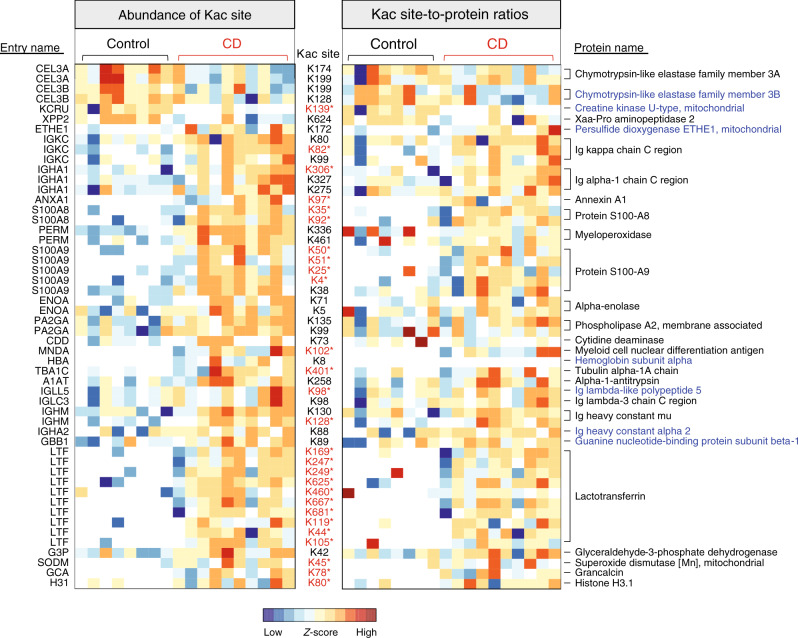
Lysine acetylation (Kac), an abundant post-translational modification (PTM) in prokaryotes, regulates various microbial metabolic pathways. However, no studies have examined protein Kac at the microbiome level, and it remains unknown whether Kac level is altered in patient microbiomes. Herein, we use a peptide immuno-affinity enrichment strategy coupled with mass spectrometry to characterize protein Kac in the microbiome, which successfully identifies 35,200 Kac peptides from microbial or human proteins in gut microbiome samples. We demonstrate that Kac is widely distributed in gut microbial metabolic pathways, including anaerobic fermentation to generate short-chain fatty acids. Applying to the analyses of microbiomes of patients with Crohn's disease identifies 52 host and 136 microbial protein Kac sites that are differentially abundant in disease versus controls. This microbiome-wide acetylomic approach aids in advancing functional microbiome research.
Conflict of interest statement
D.F., A.S., and D.M. have co-founded MedBiome, a clinical microbiomics company. C.F. and M.S. are employees of Cell Signaling Technology. The remaining authors declare no competing interests.
Figures
Similar articles
-
Global Acetylomics of Campylobacter jejuni Shows Lysine Acetylation Regulates CadF Adhesin Processing and Human Fibronectin Binding.J Proteome Res. 2023 Nov 3;22(11):3519-3533. doi: 10.1021/acs.jproteome.3c00391. Epub 2023 Oct 13. J Proteome Res. 2023. PMID: 37830485
-
Metaproteomics reveals persistent and phylum-redundant metabolic functional stability in adult human gut microbiomes of Crohn's remission patients despite temporal variations in microbial taxa, genomes, and proteomes.Microbiome. 2019 Feb 11;7(1):18. doi: 10.1186/s40168-019-0631-8. Microbiome. 2019. PMID: 30744677 Free PMC article.
-
Exploring the Microbiome-Wide Lysine Acetylation, Succinylation, and Propionylation in Human Gut Microbiota.Anal Chem. 2021 May 4;93(17):6594-6598. doi: 10.1021/acs.analchem.1c00962. Epub 2021 Apr 22. Anal Chem. 2021. PMID: 33885279
-
Protein lysine acetylation analysis: current MS-based proteomic technologies.Analyst. 2013 Mar 21;138(6):1628-36. doi: 10.1039/c3an36837h. Epub 2013 Jan 30. Analyst. 2013. PMID: 23361263 Review.
-
Dysbiotic gut microbiome: A key element of Crohn's disease.Comp Immunol Microbiol Infect Dis. 2015 Dec;43:36-49. doi: 10.1016/j.cimid.2015.10.005. Epub 2015 Oct 25. Comp Immunol Microbiol Infect Dis. 2015. PMID: 26616659 Review.
Cited by
-
Acetylated bacterial proteins as potent antigens inducing an anti-modified protein antibody response.RMD Open. 2024 Jul 22;10(3):e004411. doi: 10.1136/rmdopen-2024-004411. RMD Open. 2024. PMID: 39038910 Free PMC article.
-
Metaproteomics reveals diet-induced changes in gut microbiome function according to Crohn's disease location.Microbiome. 2024 Oct 23;12(1):217. doi: 10.1186/s40168-024-01927-5. Microbiome. 2024. PMID: 39443987 Free PMC article.
-
From risk to chronicity: evolution of autoreactive B cell and antibody responses in rheumatoid arthritis.Nat Rev Rheumatol. 2022 Jul;18(7):371-383. doi: 10.1038/s41584-022-00786-4. Epub 2022 May 23. Nat Rev Rheumatol. 2022. PMID: 35606567 Review.
-
OSaMPle workflow for salivary metaproteomics analysis reveals dysbiosis in inflammatory bowel disease patients.NPJ Biofilms Microbiomes. 2025 Apr 23;11(1):63. doi: 10.1038/s41522-025-00692-z. NPJ Biofilms Microbiomes. 2025. PMID: 40268913 Free PMC article.
-
Acetylome and Succinylome Profiling of Edwardsiella tarda Reveals Key Roles of Both Lysine Acylations in Bacterial Antibiotic Resistance.Antibiotics (Basel). 2022 Jun 23;11(7):841. doi: 10.3390/antibiotics11070841. Antibiotics (Basel). 2022. PMID: 35884095 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
