

Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics
- PMID: 32807900
- PMCID: PMC8450921
- DOI: 10.1038/s41576-020-0265-5
Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics
Abstract
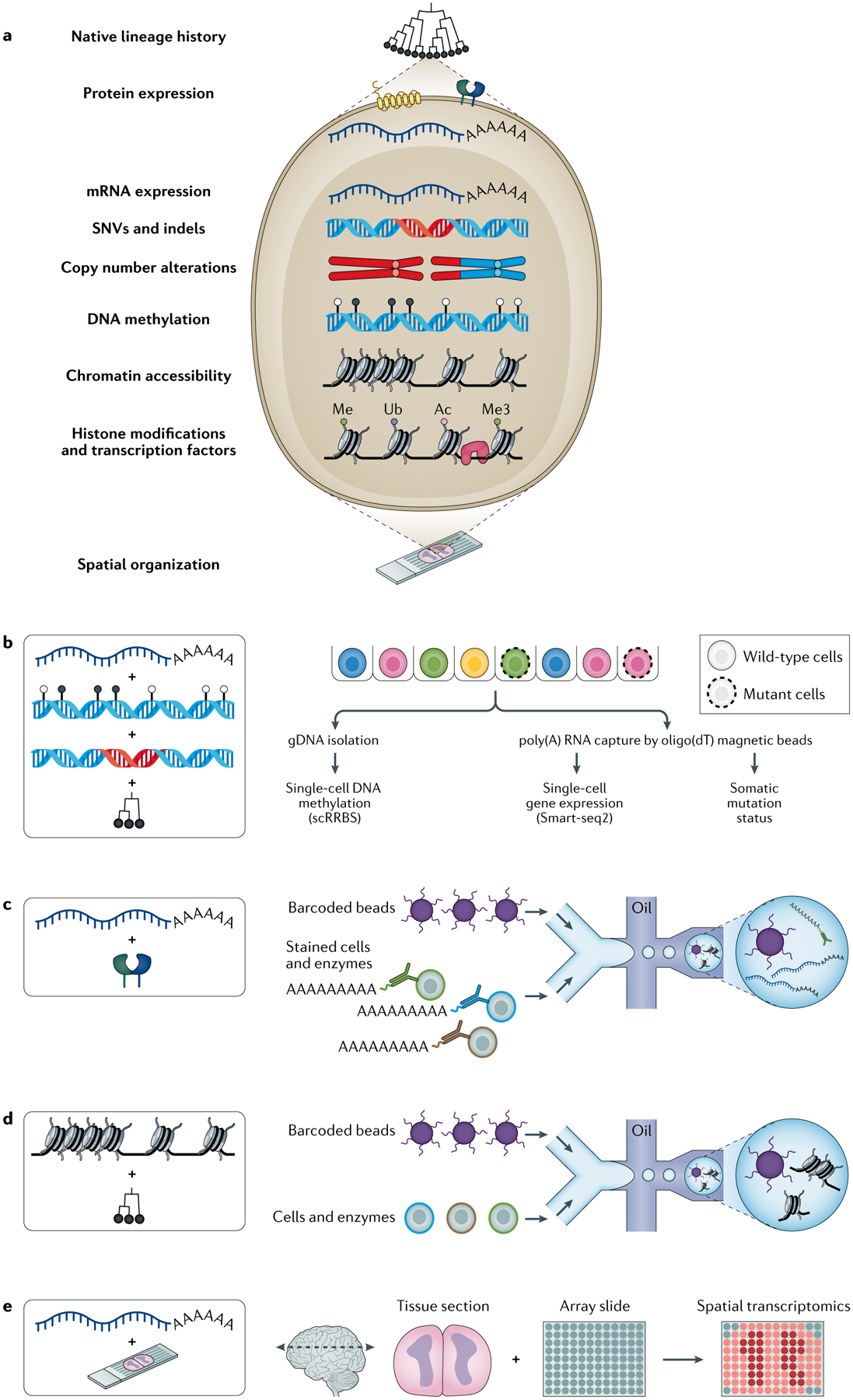
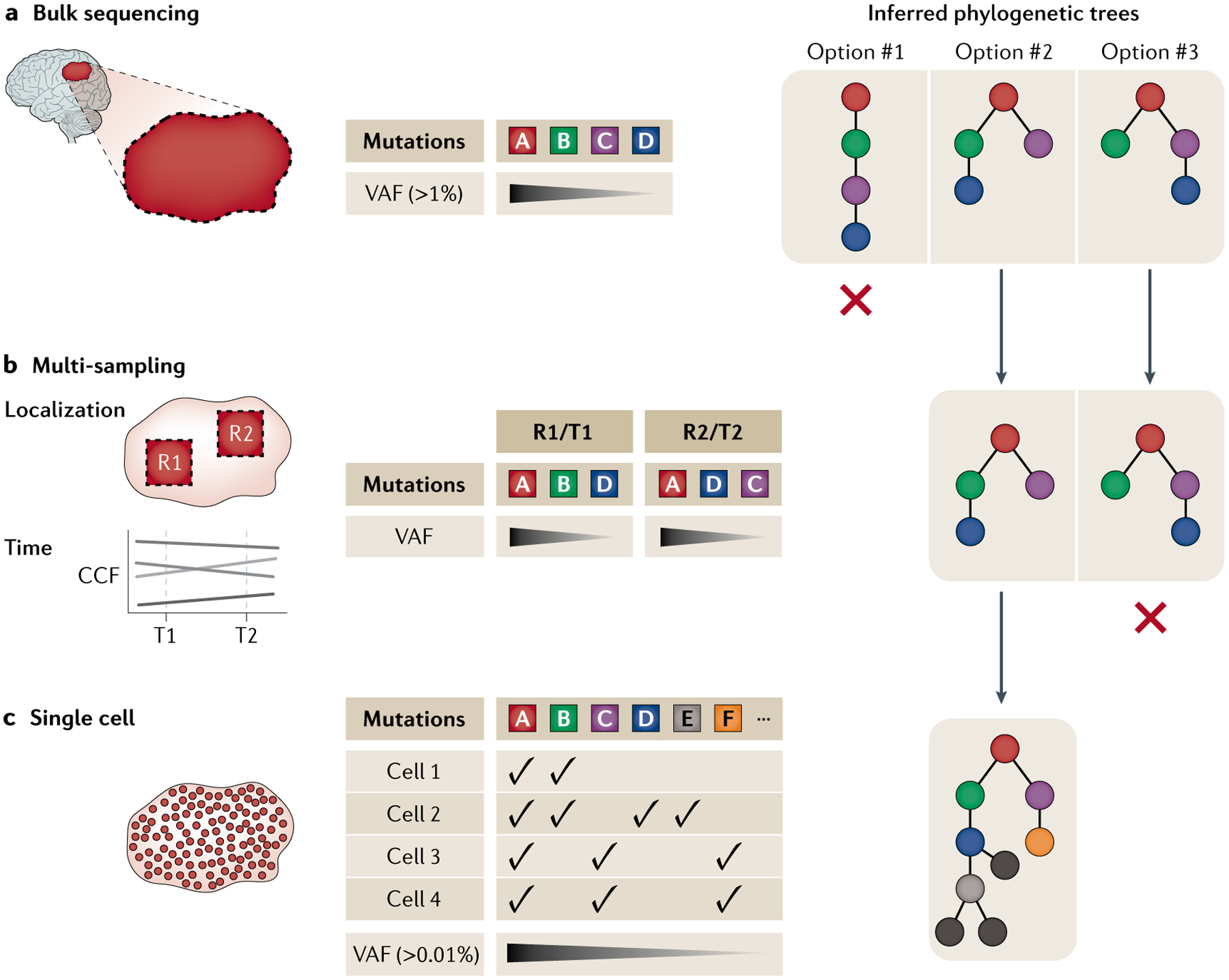
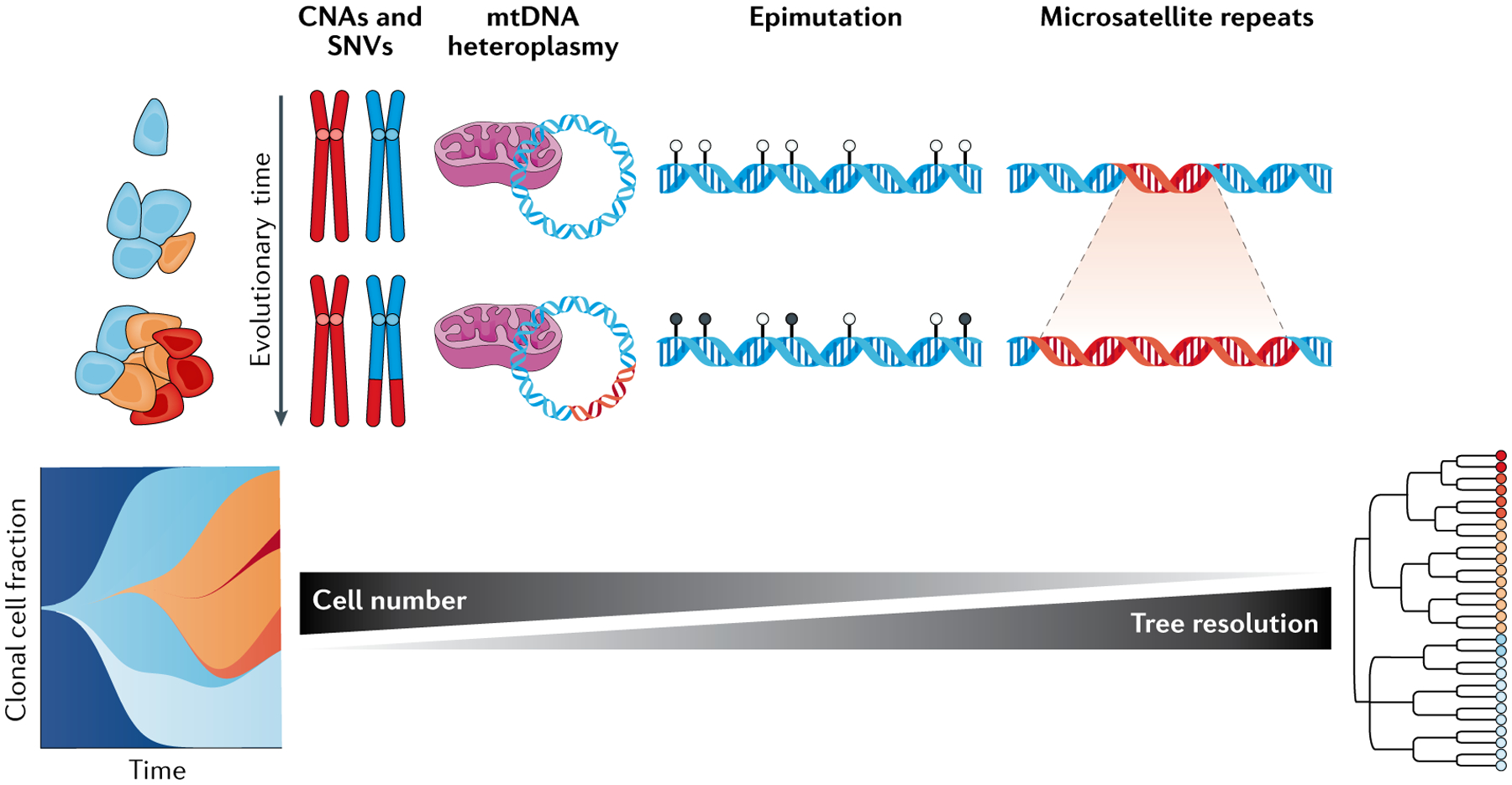
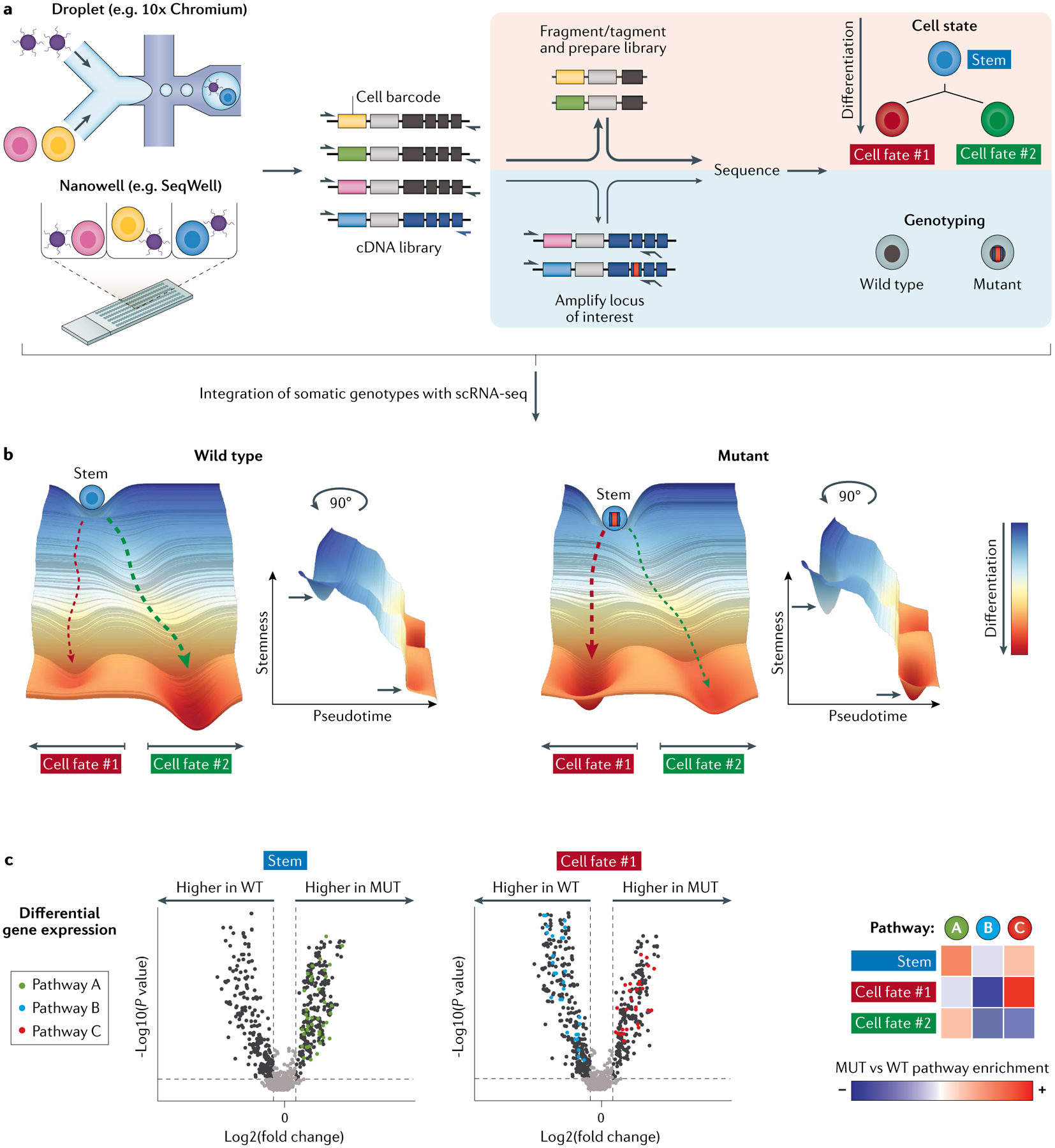
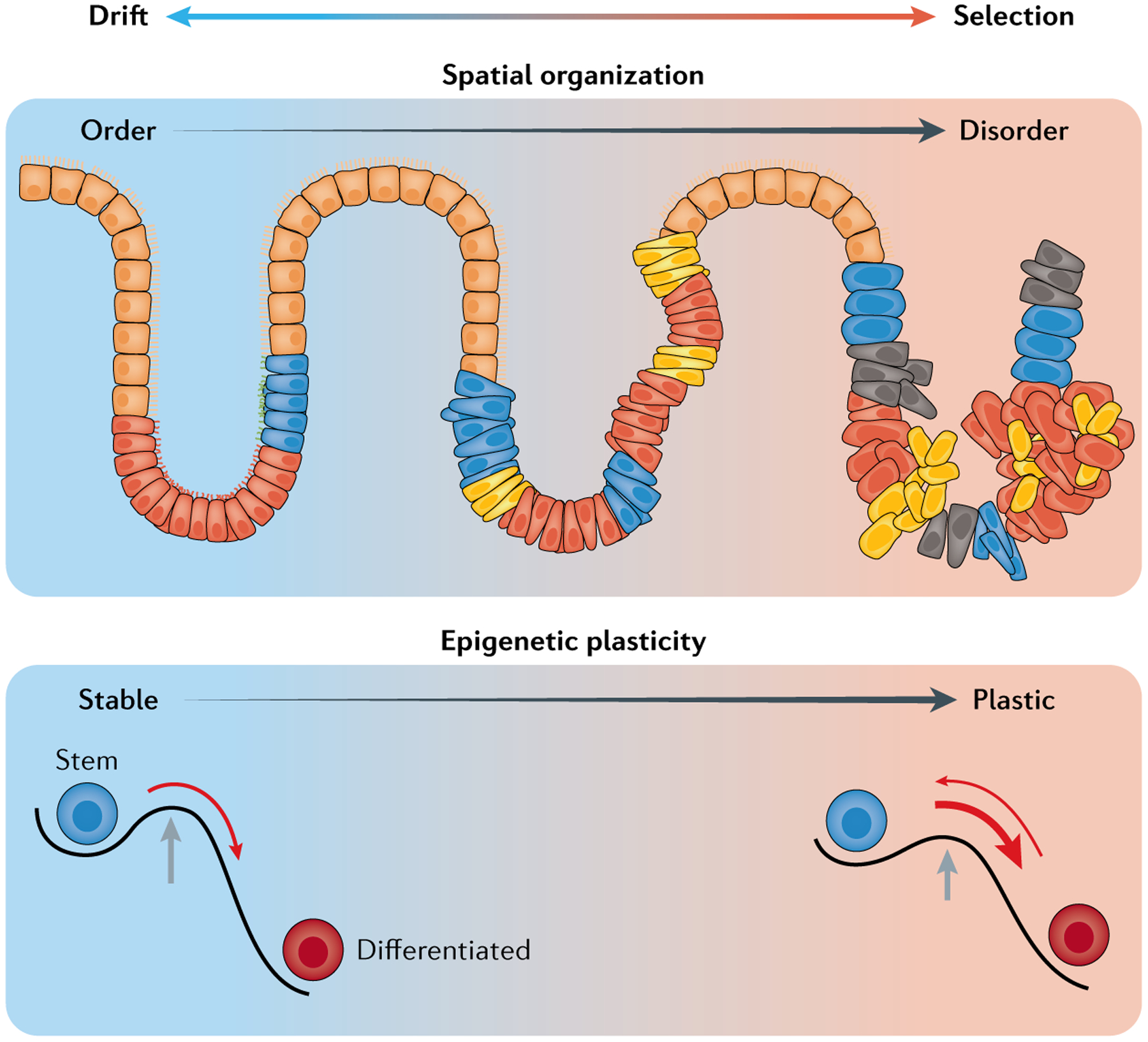
Cancer represents an evolutionary process through which growing malignant populations genetically diversify, leading to tumour progression, relapse and resistance to therapy. In addition to genetic diversity, the cell-to-cell variation that fuels evolutionary selection also manifests in cellular states, epigenetic profiles, spatial distributions and interactions with the microenvironment. Therefore, the study of cancer requires the integration of multiple heritable dimensions at the resolution of the single cell - the atomic unit of somatic evolution. In this Review, we discuss emerging analytic and experimental technologies for single-cell multi-omics that enable the capture and integration of multiple data modalities to inform the study of cancer evolution. These data show that cancer results from a complex interplay between genetic and non-genetic determinants of somatic evolution.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Turajlic S, Sottoriva A, Graham T & Swanton C Resolving genetic heterogeneity in cancer. Nat. Rev. Genet 20, 404–416 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
