

Galactokinase deficiency: lessons from the GalNet registry
- PMID: 32807972
- PMCID: PMC7790741
- DOI: 10.1038/s41436-020-00942-9
Galactokinase deficiency: lessons from the GalNet registry
Abstract
Purpose: Galactokinase (GALK1) deficiency is a rare hereditary galactose metabolism disorder. Beyond cataract, the phenotypic spectrum is questionable. Data from affected patients included in the Galactosemias Network registry were collected to better characterize the phenotype.
Methods: Observational study collecting medical data of 53 not previously reported GALK1 deficient patients from 17 centers in 11 countries from December 2014 to April 2020.
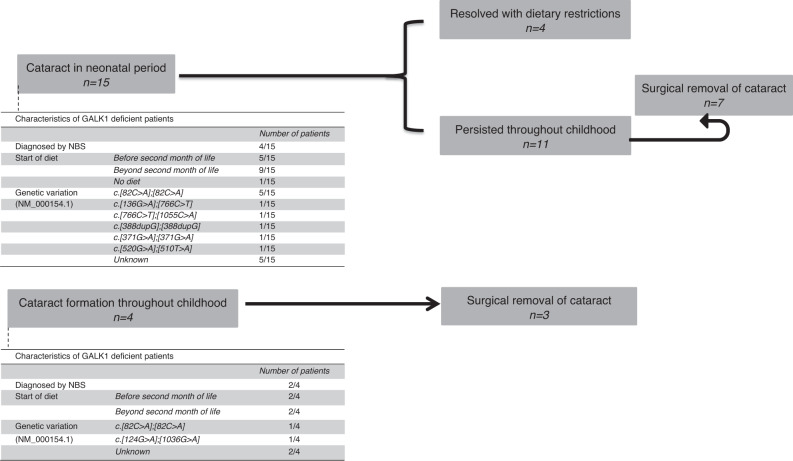
Results: Neonatal or childhood cataract was reported in 15 and 4 patients respectively. The occurrence of neonatal hypoglycemia and infection were comparable with the general population, whereas bleeding diathesis (8.1% versus 2.17-5.9%) and encephalopathy (3.9% versus 0.3%) were reported more often. Elevated transaminases were seen in 25.5%. Cognitive delay was reported in 5 patients. Urinary galactitol was elevated in all patients at diagnosis; five showed unexpected Gal-1-P increase. Most patients showed enzyme activities ≤1%. Eleven different genotypes were described, including six unpublished variants. The majority was homozygous for NM_000154.1:c.82C>A (p.Pro28Thr). Thirty-five patients were diagnosed following newborn screening, which was clearly beneficial.
Conclusion: The phenotype of GALK1 deficiency may include neonatal elevation of transaminases, bleeding diathesis, and encephalopathy in addition to cataract. Potential complications beyond the neonatal period are not systematically surveyed and a better delineation is needed.
Keywords: GALK1 gene variants; neonatal complications; cataract; galactosemias registry; galactokinase 1 deficiency.
Conflict of interest statement
A.M.B. has been a member of advisory boards for Nutricia and Biomarin and has received a speaking fee. The other authors declare no conflicts of interest.
Figures
References
-
- Fanconi G. Marked galactose intolerance (galactose diabetes) in a child with neuronbromatosis Eecklinghausen. Jahrb Kinderheilkd. 1933;138:1–8.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
