

Rare genetic causes of complex kidney and urological diseases
- PMID: 32807983
- PMCID: PMC7772719
- DOI: 10.1038/s41581-020-0325-2
Rare genetic causes of complex kidney and urological diseases
Abstract
Although often considered a single-entity, chronic kidney disease (CKD) comprises many pathophysiologically distinct disorders that result in persistently abnormal kidney structure and/or function, and encompass both monogenic and polygenic aetiologies. Rare inherited forms of CKD frequently span diverse phenotypes, reflecting genetic phenomena including pleiotropy, incomplete penetrance and variable expressivity. Use of chromosomal microarray and massively parallel sequencing technologies has revealed that genomic disorders and monogenic aetiologies contribute meaningfully to seemingly complex forms of CKD across different clinically defined subgroups and are characterized by high genetic and phenotypic heterogeneity. Investigations of prevalent genomic disorders in CKD have integrated genetic, bioinformatic and functional studies to pinpoint the genetic drivers underlying their renal and extra-renal manifestations, revealing both monogenic and polygenic mechanisms. Similarly, massively parallel sequencing-based analyses have identified gene- and allele-level variation that contribute to the clinically diverse phenotypes observed for many monogenic forms of nephropathy. Genome-wide sequencing studies suggest that dual genetic diagnoses are found in at least 5% of patients in whom a genetic cause of disease is identified, highlighting the fact that complex phenotypes can also arise from multilocus variation. A multifaceted approach that incorporates genetic and phenotypic data from large, diverse cohorts will help to elucidate the complex relationships between genotype and phenotype for different forms of CKD, supporting personalized medicine for individuals with kidney disease.
Figures
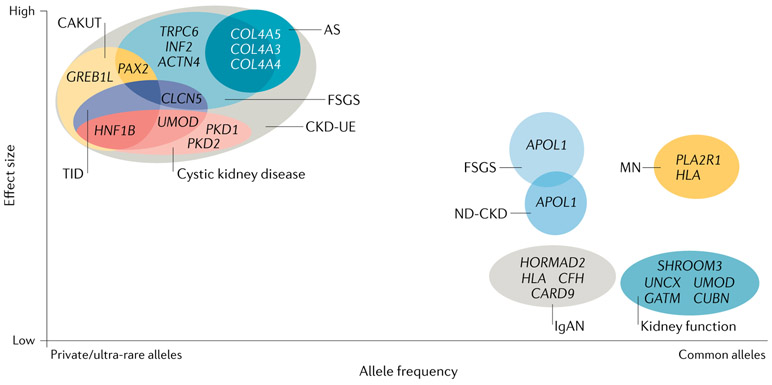
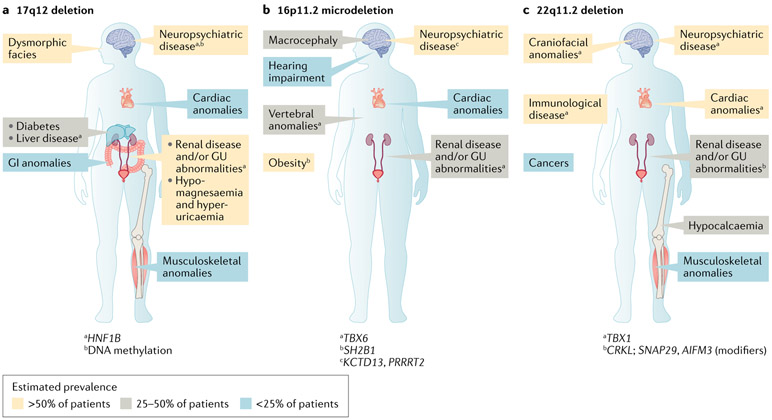
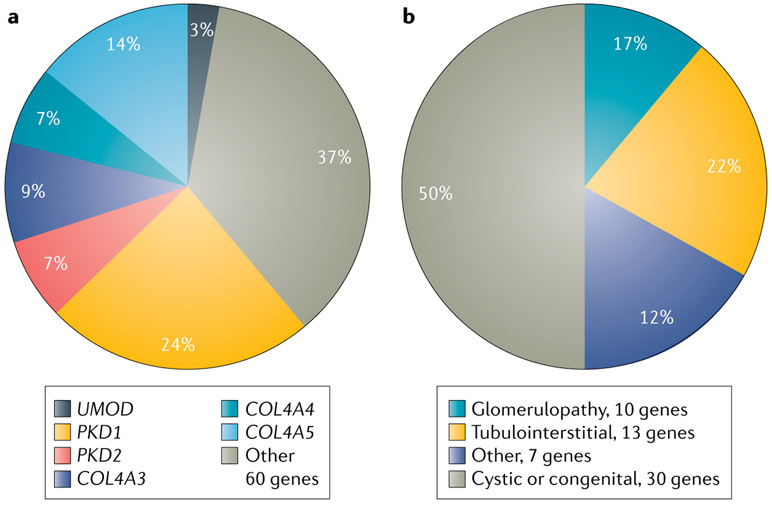
References
-
- Webster AC, Nagler EV, Morton RL & Masson P Chronic kidney disease. Lancet 389, 1238–1252 (2017). - PubMed
-
- Connaughton DM et al. The Irish kidney gene project-prevalence of family history in patients with kidney disease in ireland. Nephron 130, 293–301 (2015). - PubMed
-
- McClellan WM et al. Individuals with a family history of ESRD are a high-risk population for CKD: implications for targeted surveillance and intervention activities. Am. J. Kidney Dis 53, S100–S106 (2009). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
