

Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers
- PMID: 32807987
- PMCID: PMC7484012
- DOI: 10.1038/s41588-020-0678-2
Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers
Abstract
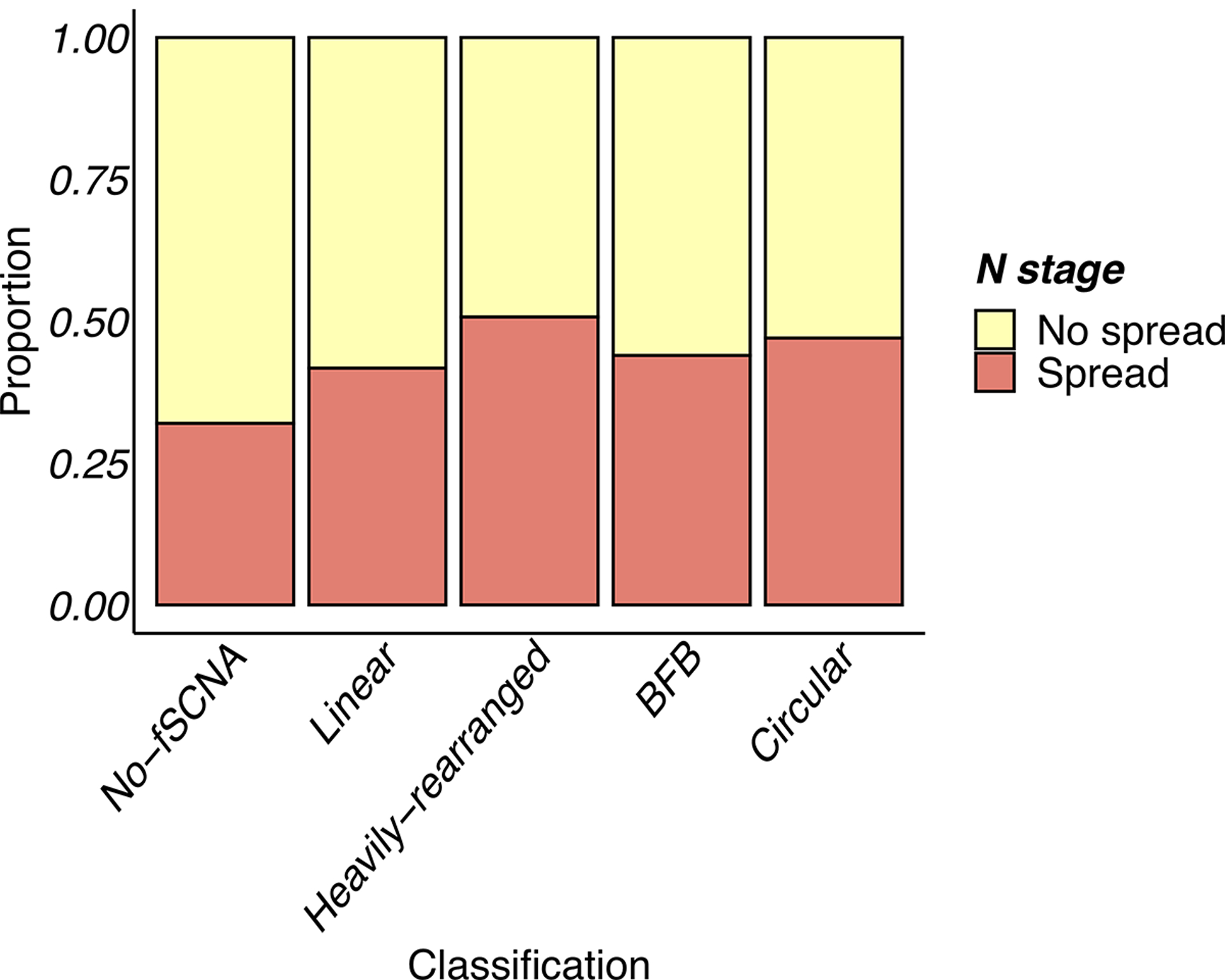
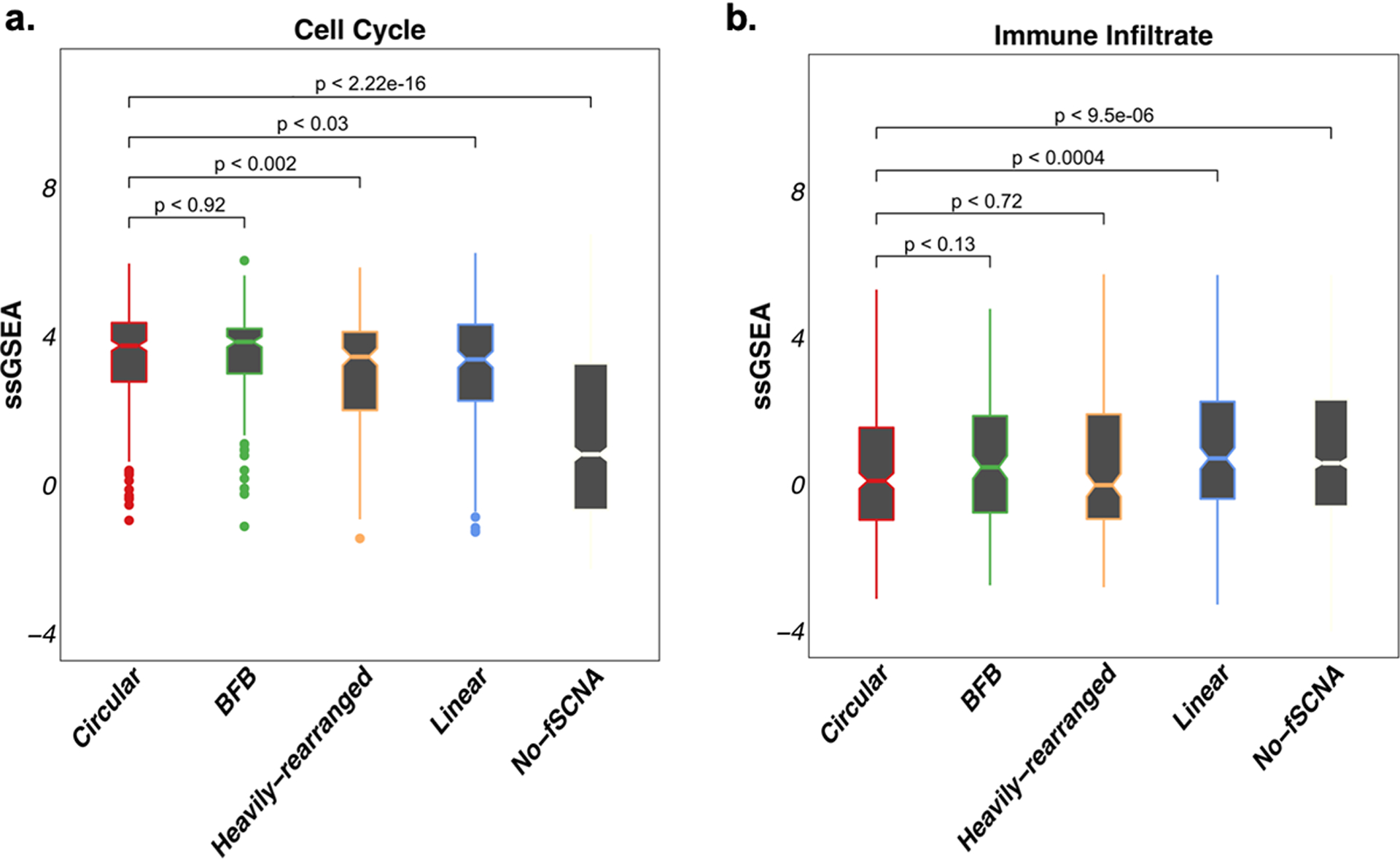
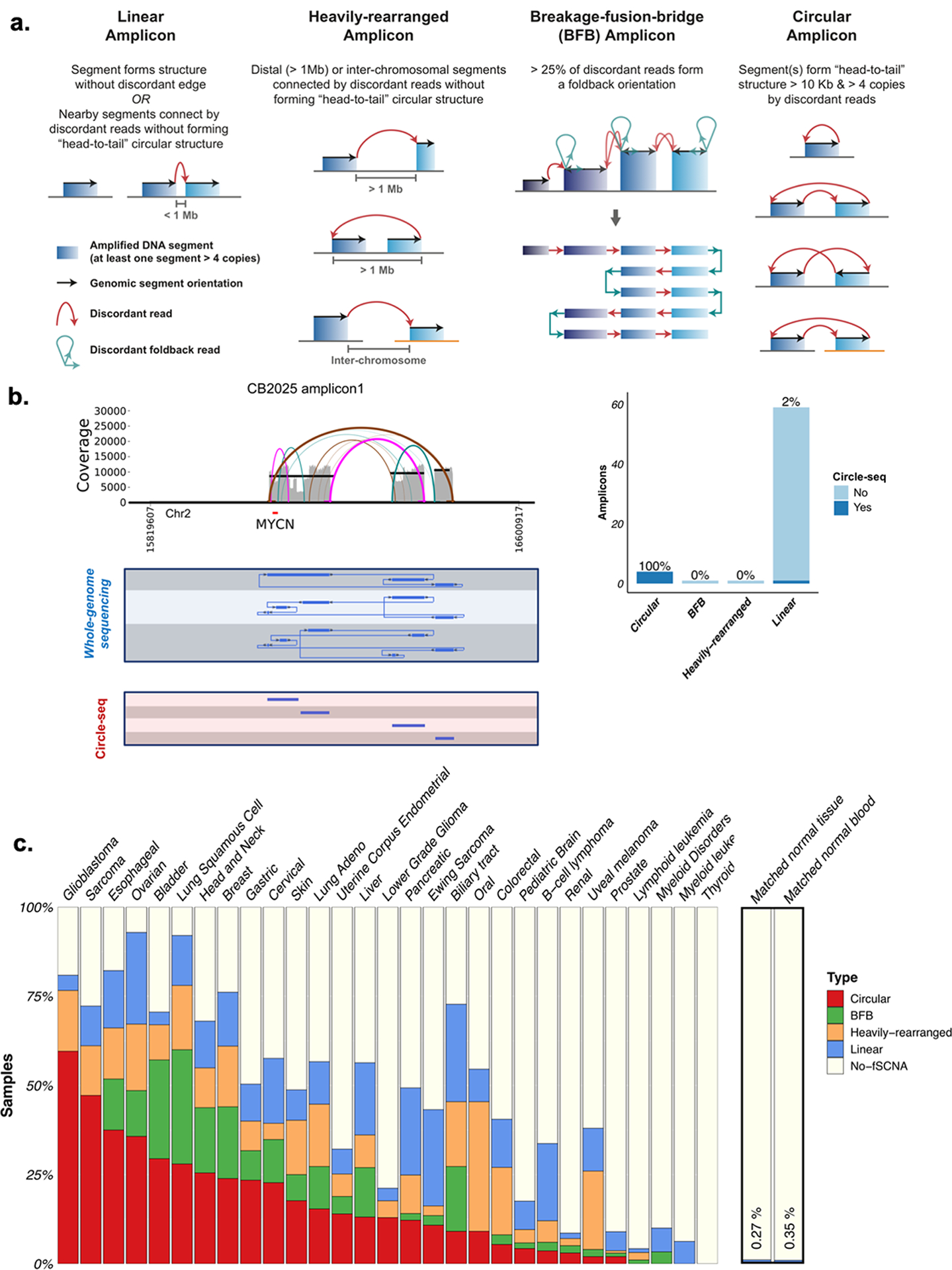
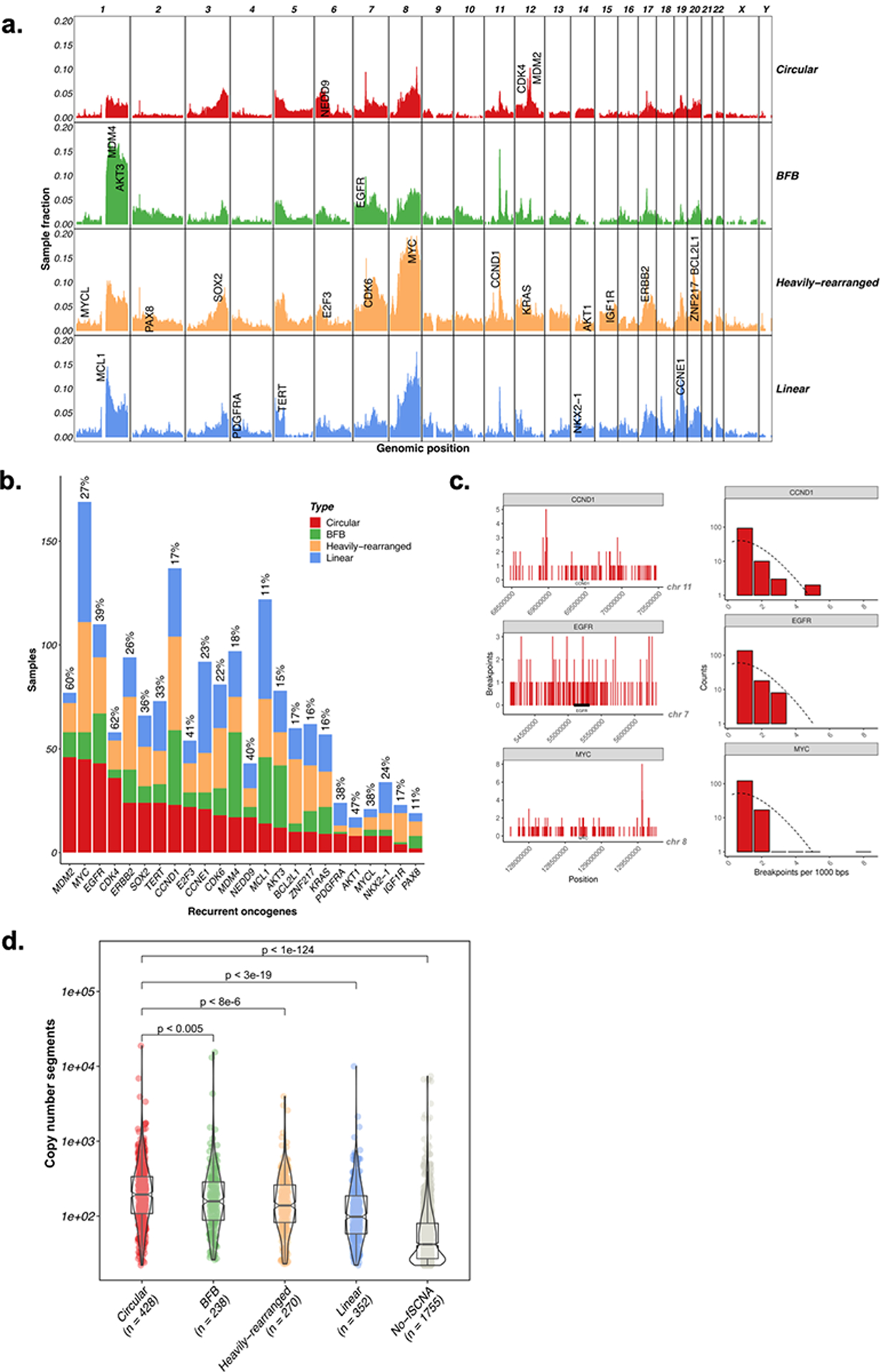
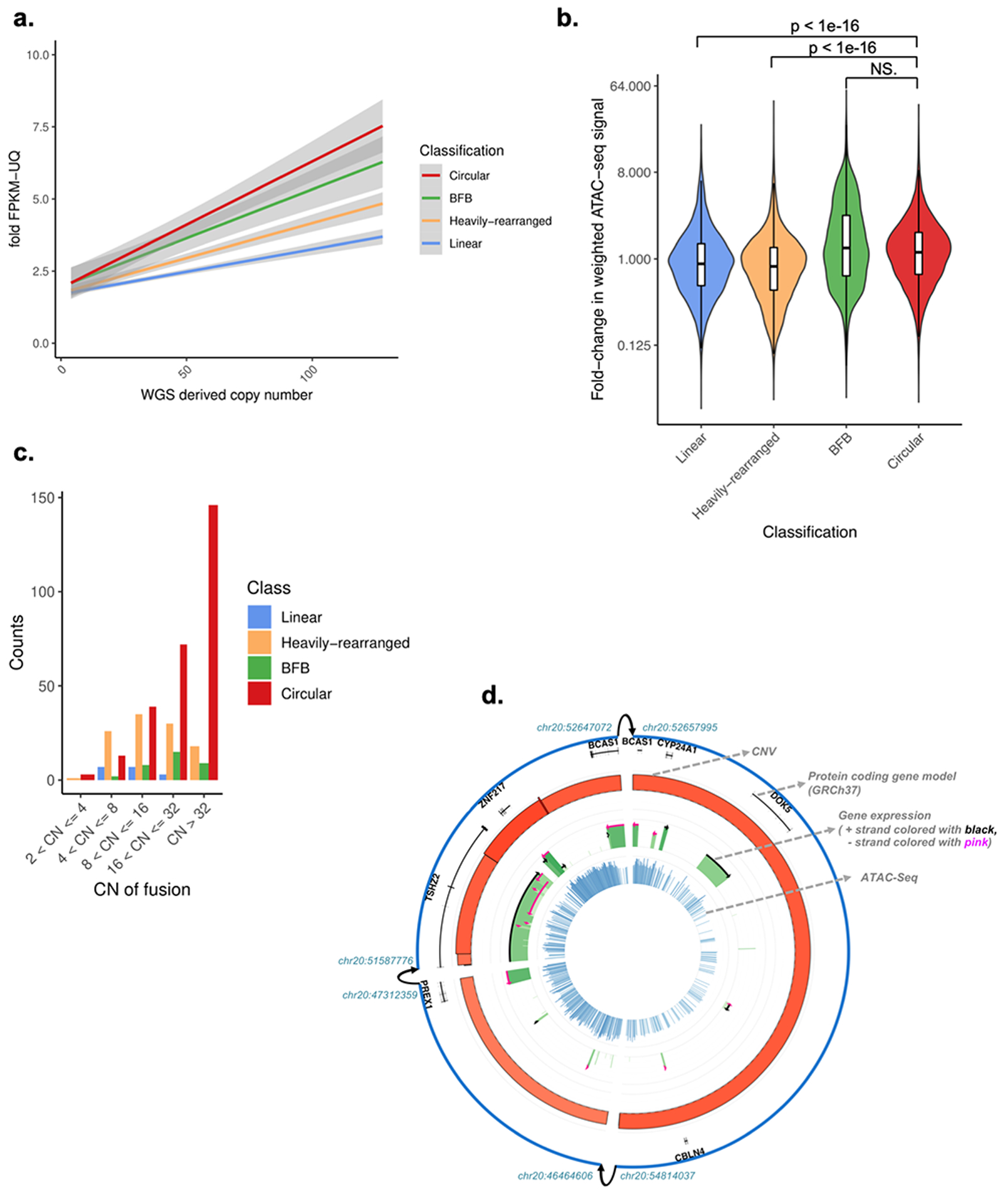
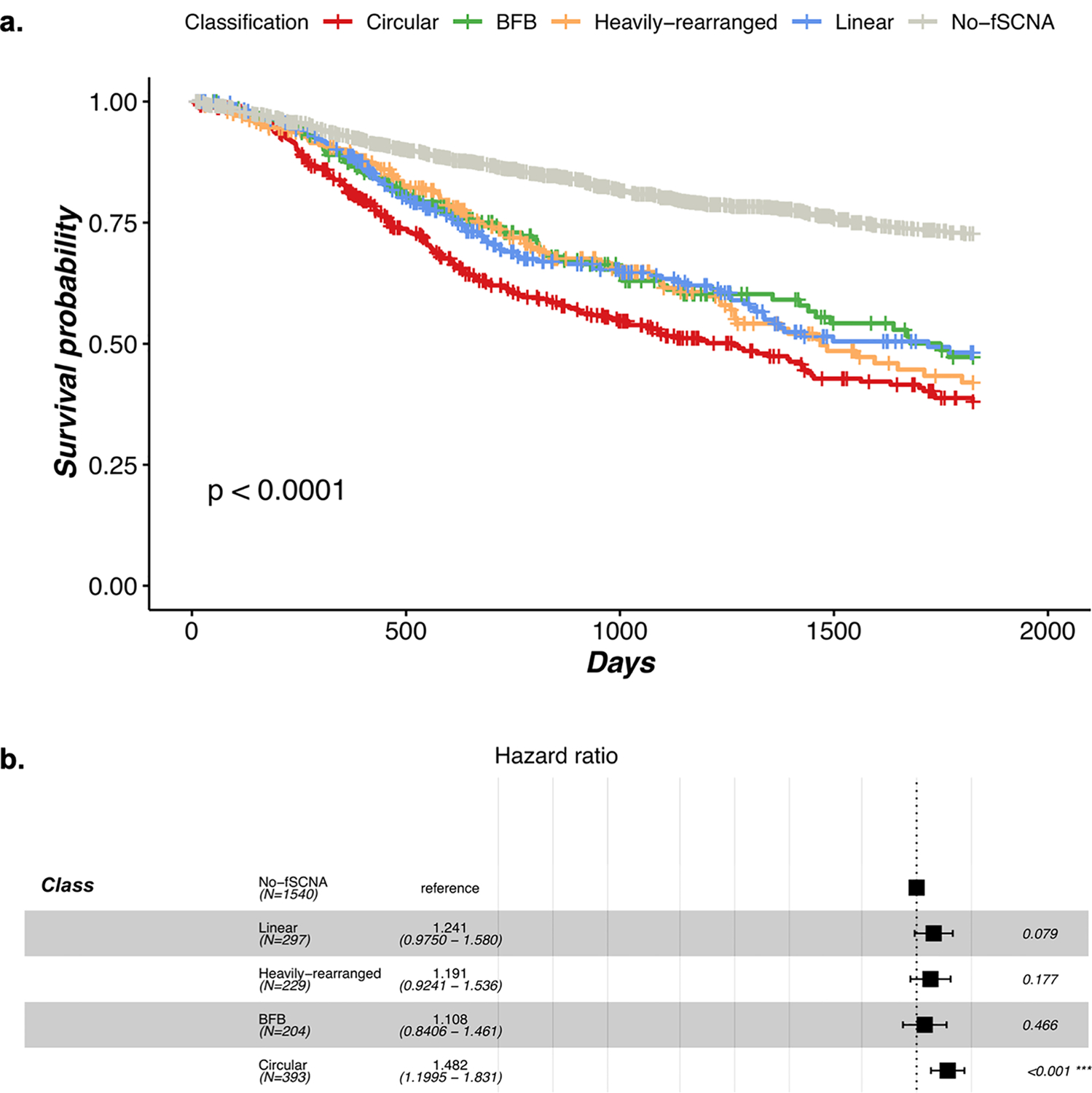
Extrachromosomal DNA (ecDNA) amplification promotes intratumoral genetic heterogeneity and accelerated tumor evolution1-3; however, its frequency and clinical impact are unclear. Using computational analysis of whole-genome sequencing data from 3,212 cancer patients, we show that ecDNA amplification frequently occurs in most cancer types but not in blood or normal tissue. Oncogenes were highly enriched on amplified ecDNA, and the most common recurrent oncogene amplifications arose on ecDNA. EcDNA amplifications resulted in higher levels of oncogene transcription compared to copy number-matched linear DNA, coupled with enhanced chromatin accessibility, and more frequently resulted in transcript fusions. Patients whose cancers carried ecDNA had significantly shorter survival, even when controlled for tissue type, than patients whose cancers were not driven by ecDNA-based oncogene amplification. The results presented here demonstrate that ecDNA-based oncogene amplification is common in cancer, is different from chromosomal amplification and drives poor outcome for patients across many cancer types.
Conflict of interest statement
COMPETING INTERESTS
H.Y.C., P.S.M., V.B. and R.G.W.V. are scientific co-founders of Boundless Bio, Inc. (BBI), and serve as consultants. V.B. is a co-founder, and has equity interest in Digital Proteomics, LLC (DP), and receives income from DP. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. N.P. N. and K.T. are employees of Boundless Bio, Inc.
Figures
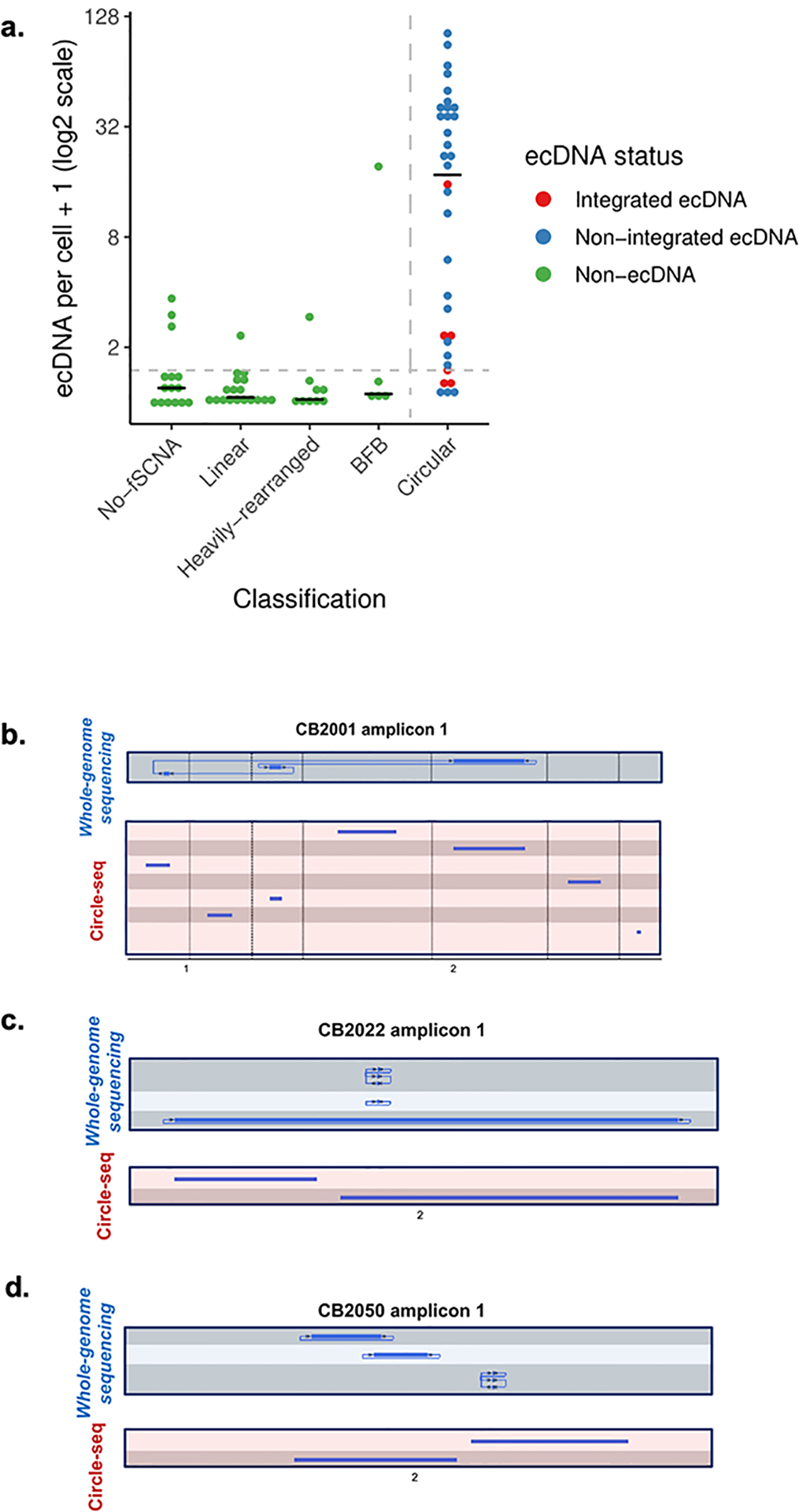
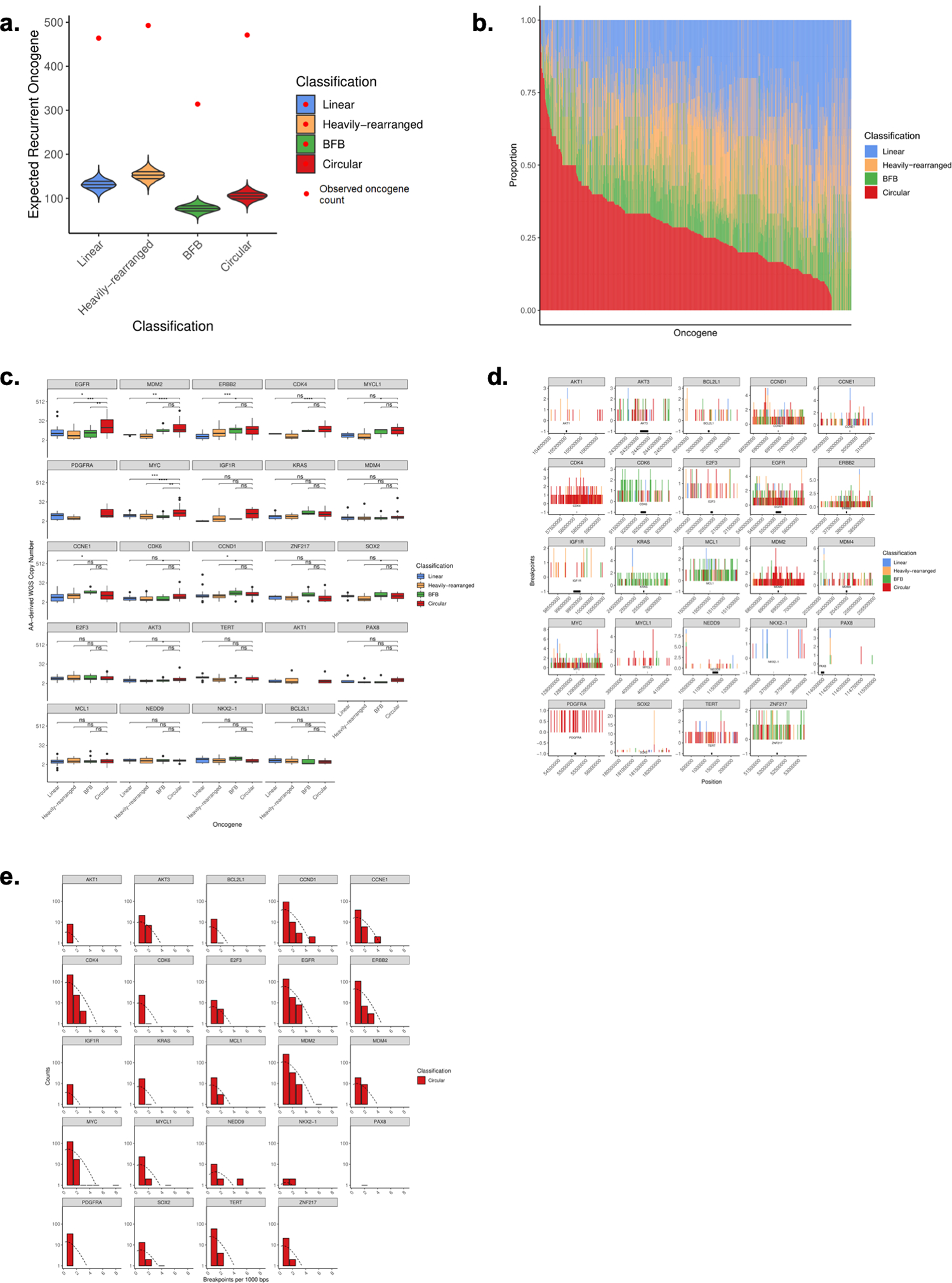
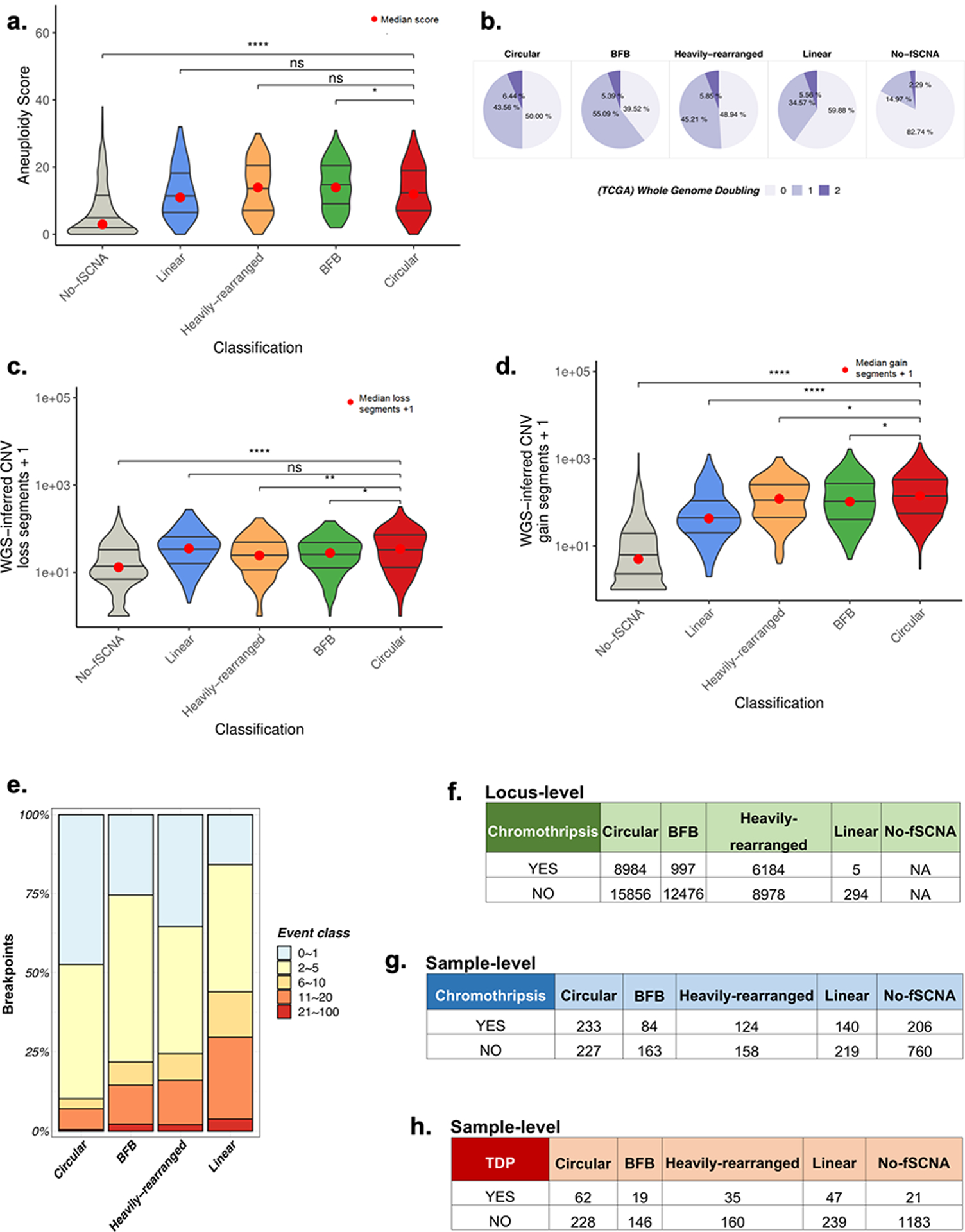
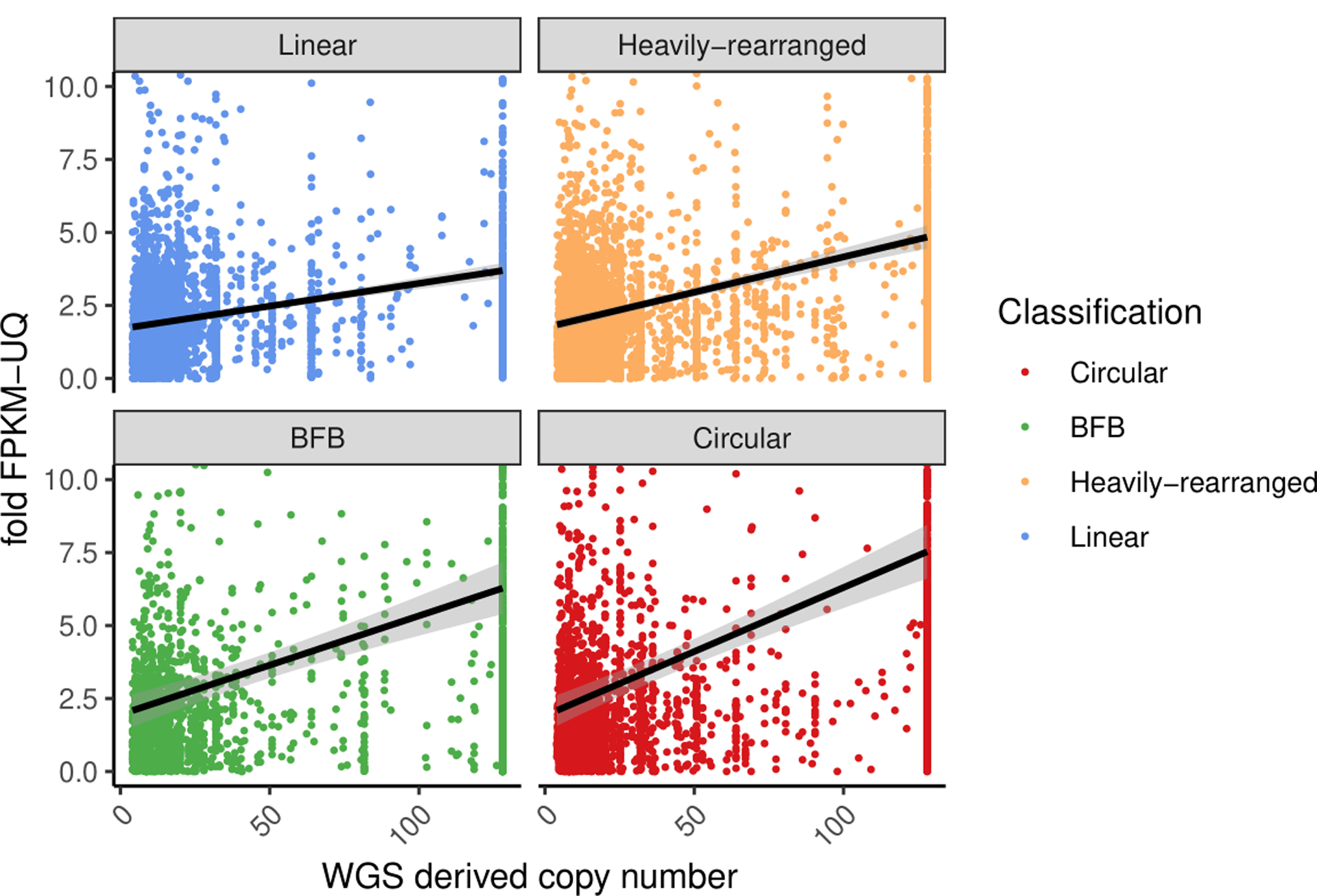
Comment in
-
ecDNA within tumors: a new mechanism that drives tumor heterogeneity and drug resistance.Signal Transduct Target Ther. 2020 Nov 24;5(1):277. doi: 10.1038/s41392-020-00403-4. Signal Transduct Target Ther. 2020. PMID: 33235201 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R01 CA190121/CA/NCI NIH HHS/United States
- R21 NS114873/NS/NINDS NIH HHS/United States
- R35 GM133600/GM/NIGMS NIH HHS/United States
- R01 NS073831/NS/NINDS NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- R01 CA237208/CA/NCI NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- R01 GM114362/GM/NIGMS NIH HHS/United States
- R35 CA209919/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P30 CA034196/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
