

Lyso-glycosphingolipids: presence and consequences
- PMID: 32808655
- PMCID: PMC7517347
- DOI: 10.1042/EBC20190090
Lyso-glycosphingolipids: presence and consequences
Abstract
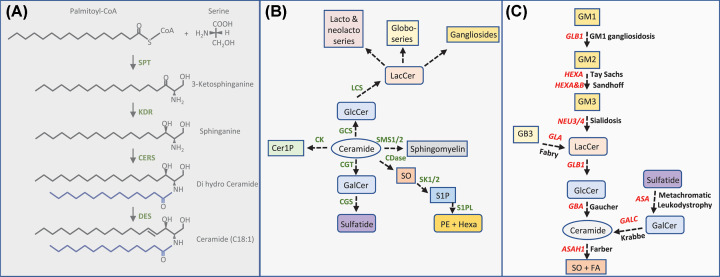
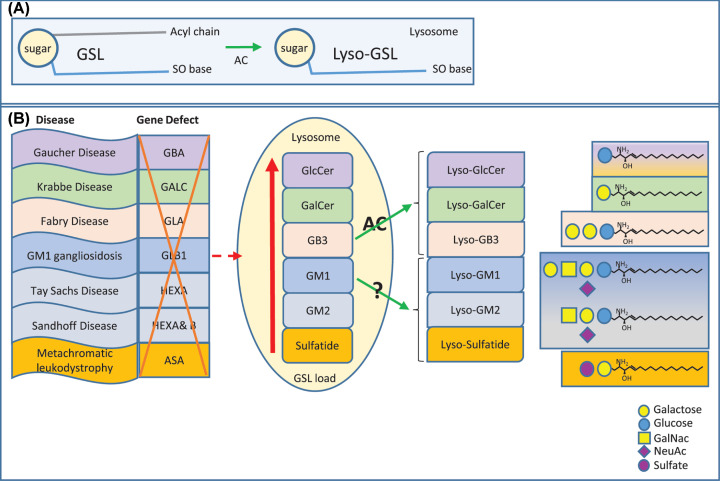
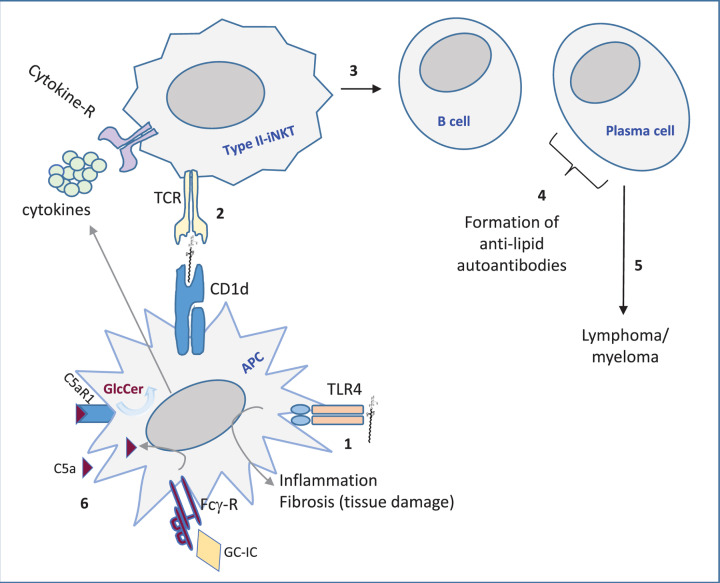
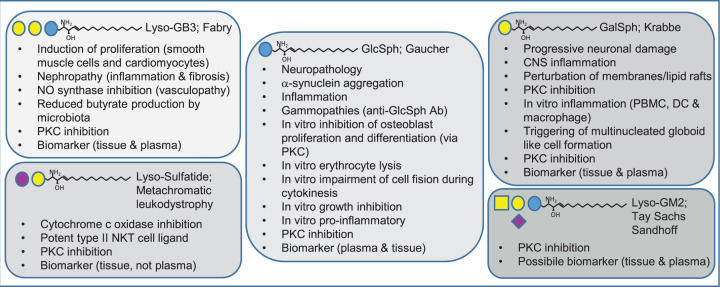
Lyso-glycosphingolipids are generated in excess in glycosphingolipid storage disorders. In the course of these pathologies glycosylated sphingolipid species accumulate within lysosomes due to flaws in the respective lipid degrading machinery. Deacylation of accumulating glycosphingolipids drives the formation of lyso-glycosphingolipids. In lysosomal storage diseases such as Gaucher Disease, Fabry Disease, Krabbe disease, GM1 -and GM2 gangliosidosis, Niemann Pick type C and Metachromatic leukodystrophy massive intra-lysosomal glycosphingolipid accumulation occurs. The lysosomal enzyme acid ceramidase generates the deacylated lyso-glycosphingolipid species. This review discusses how the various lyso-glycosphingolipids are synthesized, how they may contribute to abnormal immunity in glycosphingolipid storing lysosomal diseases and what therapeutic opportunities exist.
Keywords: acid ceramidase; glycosphingolipid; immune response; lysosomal storage disease.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
References
-
- Thudichum J. (1884) A Treatise on the Chemical constitution of the brain, Bailliere, Tindall and Cox., London
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical