

Spinal CCL2 Promotes Pain Sensitization by Rapid Enhancement of NMDA-Induced Currents Through the ERK-GluN2B Pathway in Mouse Lamina II Neurons
- PMID: 32809188
- PMCID: PMC7674543
- DOI: 10.1007/s12264-020-00557-9
Spinal CCL2 Promotes Pain Sensitization by Rapid Enhancement of NMDA-Induced Currents Through the ERK-GluN2B Pathway in Mouse Lamina II Neurons
Abstract
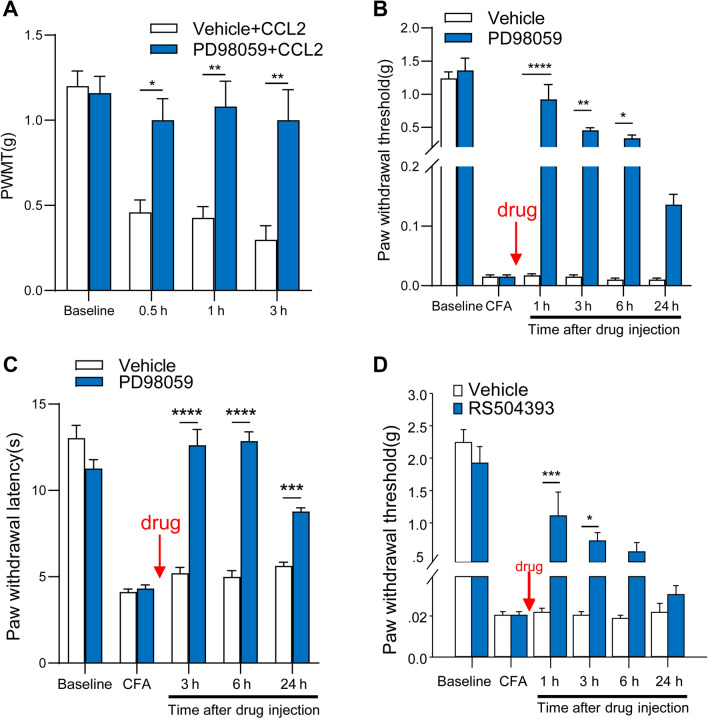
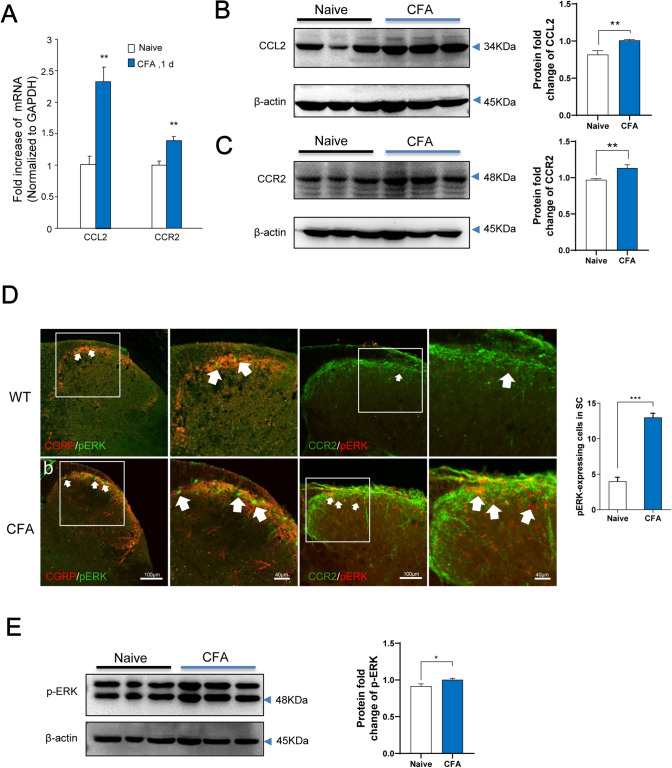
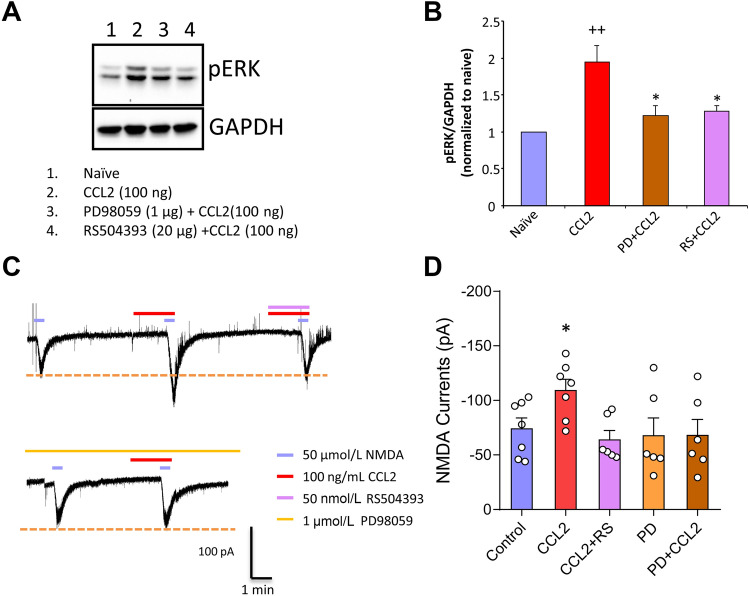
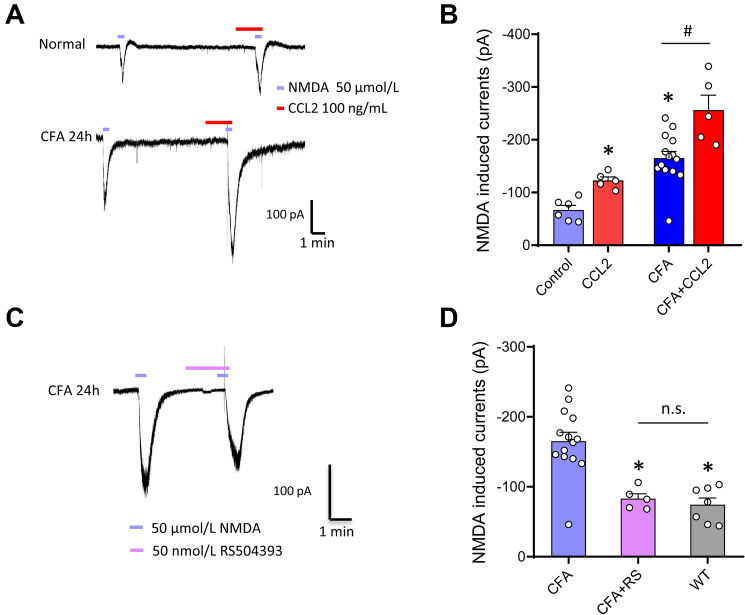
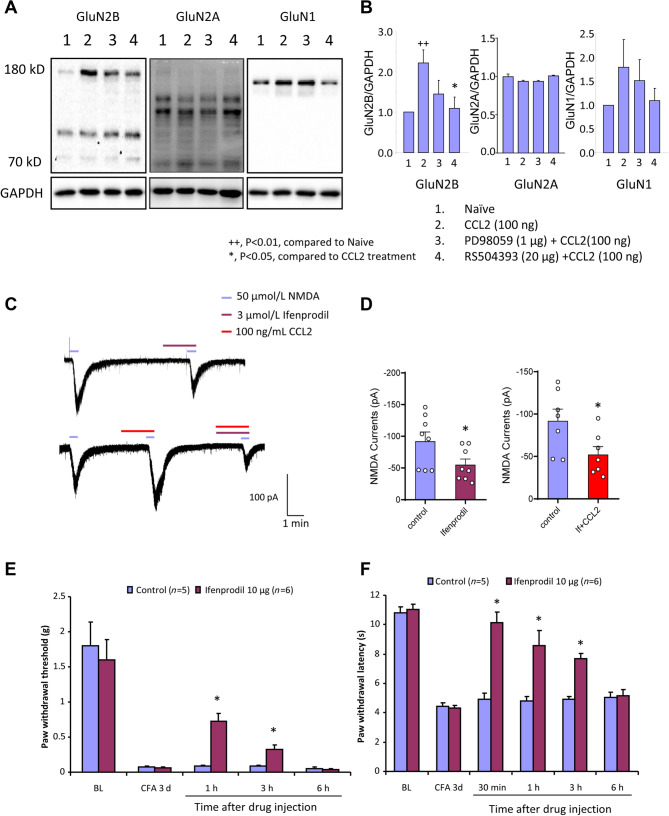
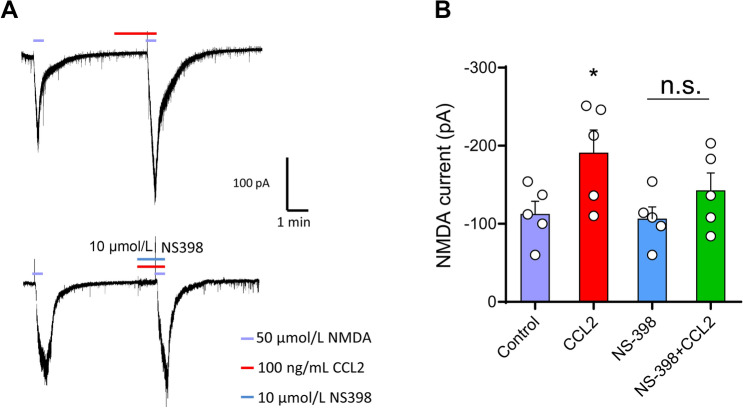
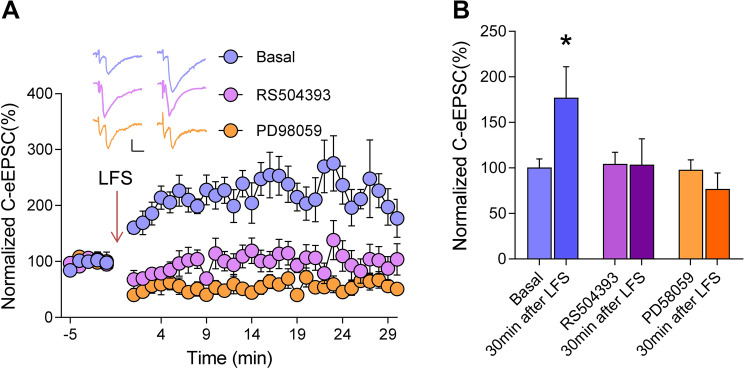
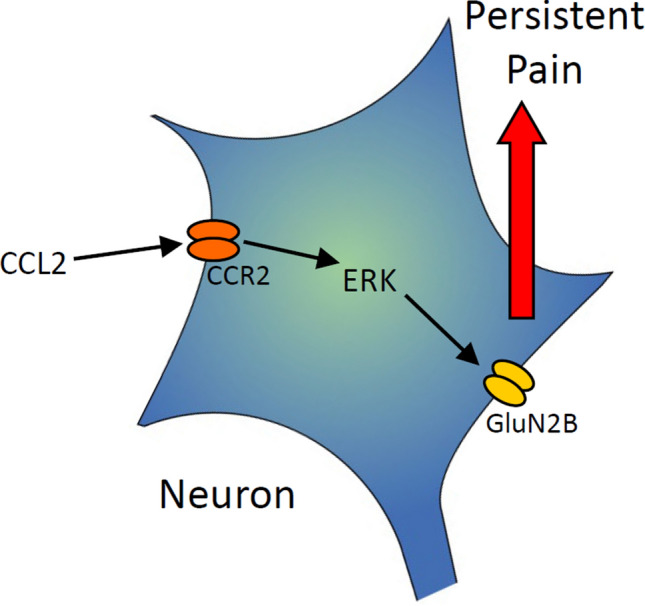
Previous studies have shown that CCL2 (C-C motif chemokine ligand 2) induces chronic pain, but the exact mechanisms are still unknown. Here, we established models to explore the potential mechanisms. Behavioral experiments revealed that an antagonist of extracellular signal-regulated kinase (ERK) inhibited not only CCL2-induced inflammatory pain, but also pain responses induced by complete Freund's adjuvant. We posed the question of the intracellular signaling cascade involved. Subsequent experiments showed that CCL2 up-regulated the expression of phosphorylated ERK (pERK) and N-methyl D-aspartate receptor [NMDAR] subtype 2B (GluN2B); meanwhile, antagonists of CCR2 and ERK effectively reversed these phenomena. Whole-cell patch-clamp recordings revealed that CCL2 enhanced the NMDAR-induced currents via activating the pERK pathway, which was blocked by antagonists of GluN2B and ERK. In summary, we demonstrate that CCL2 directly interacts with CCR2 to enhance NMDAR-induced currents, eventually leading to inflammatory pain mainly through the CCL2-CCR2-pERK-GluN2B pathway.
Keywords: C-C motif chemokine ligand 2; Extracellular signal-regulated kinase; Monocyte chemoattractant protein 1; Neuron-glial interaction.
Conflict of interest statement
The authors claim that there are no conflicts of interest.
Figures
References
-
- Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12:55–61. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
