

Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome
- PMID: 32810435
- PMCID: PMC7541479
- DOI: 10.1016/j.stem.2020.07.015
Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome
Abstract
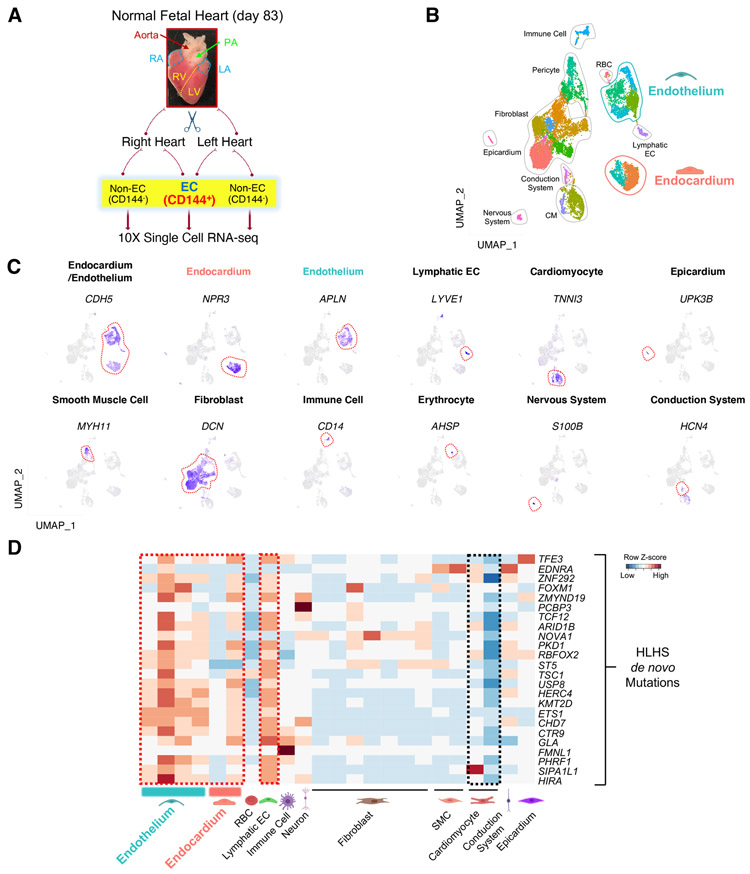
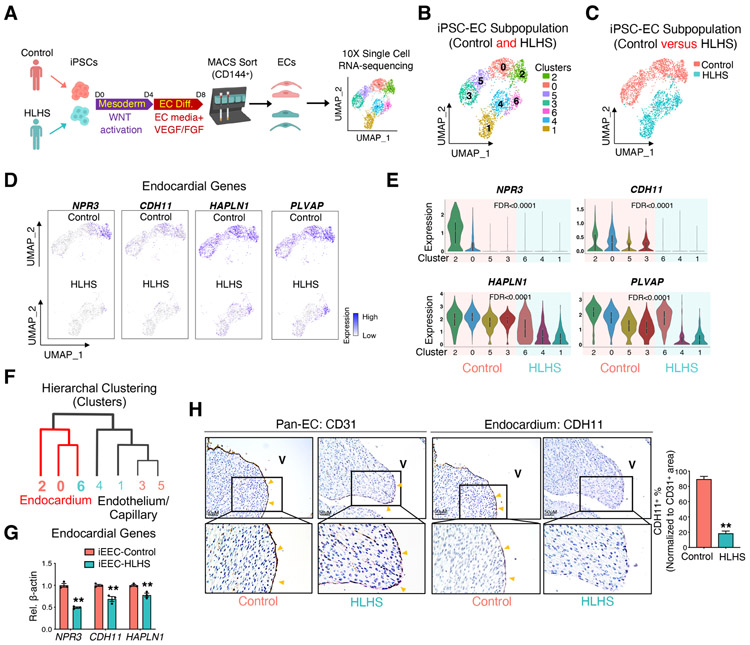
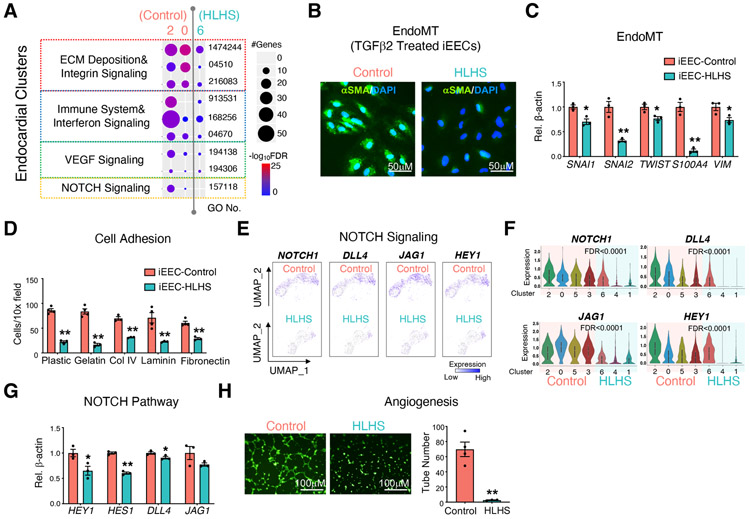
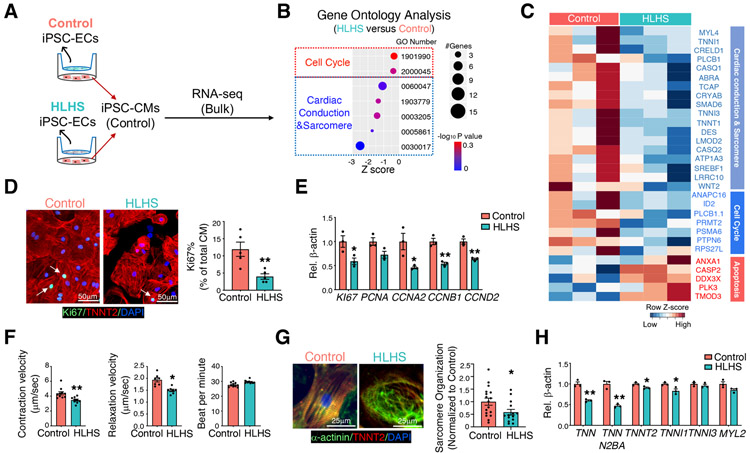
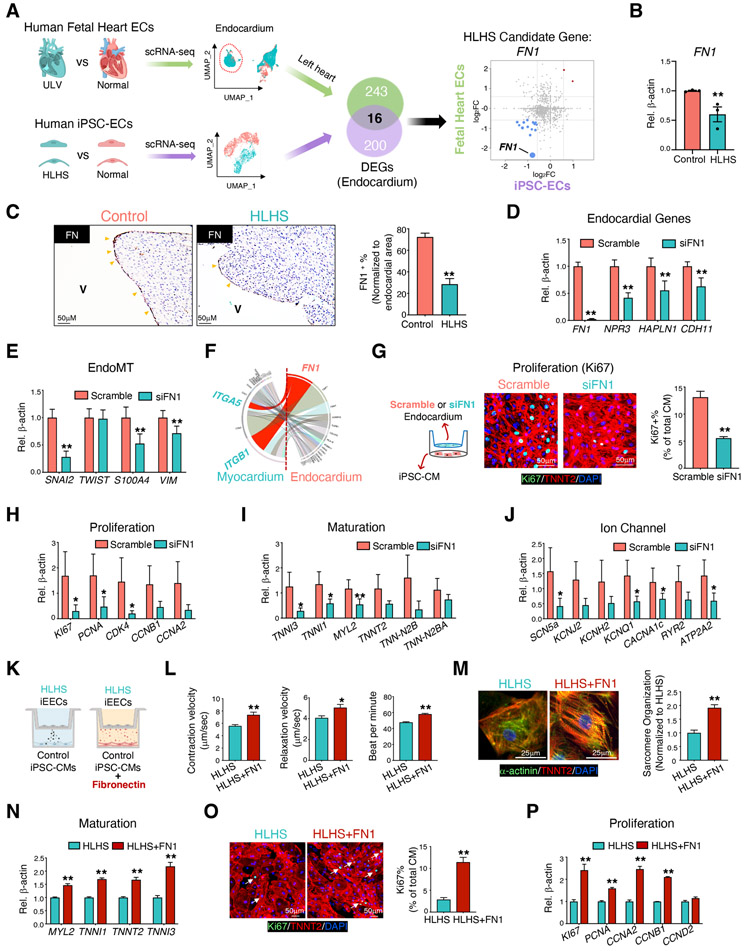
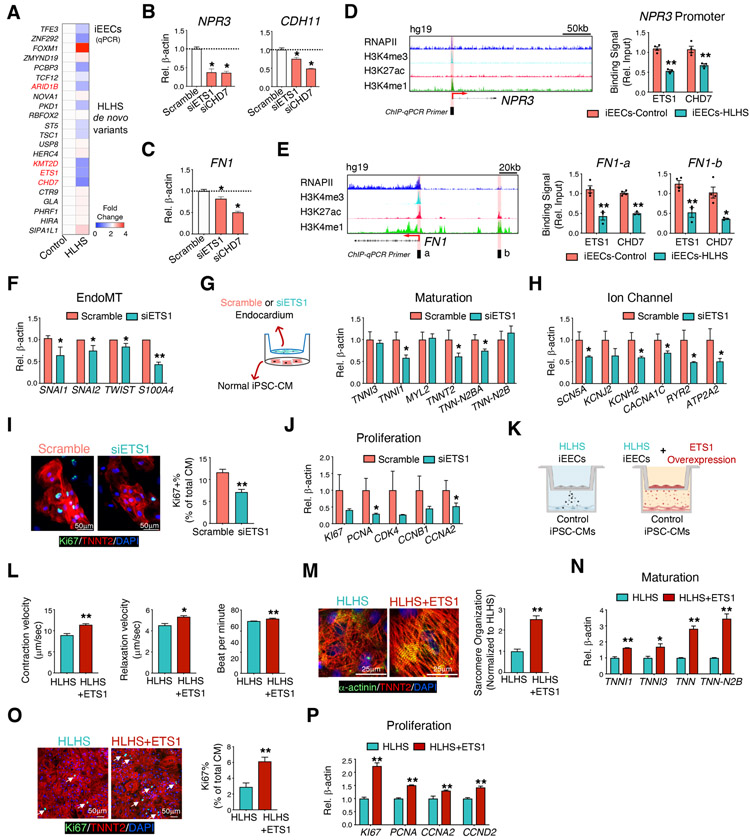
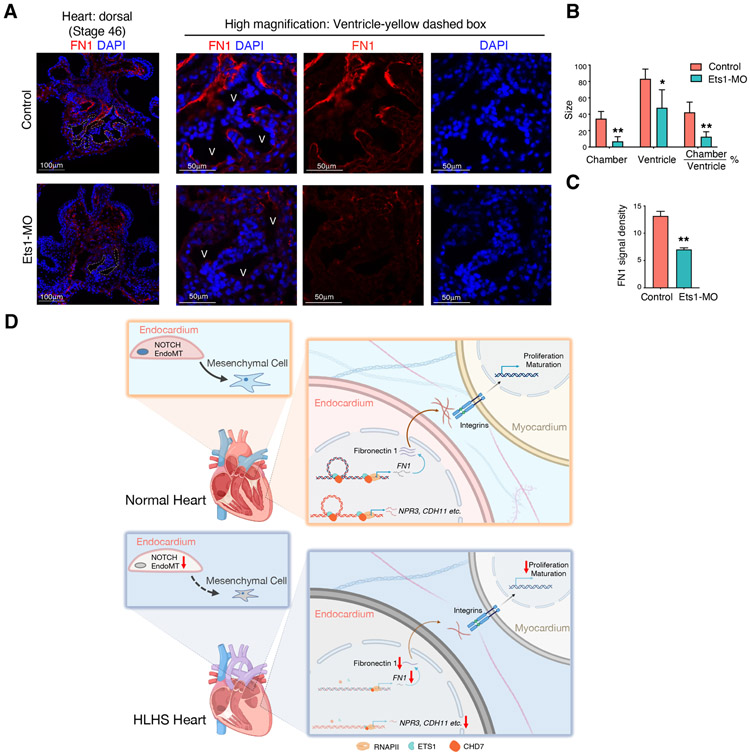
Hypoplastic left heart syndrome (HLHS) is a complex congenital heart disease characterized by abnormalities in the left ventricle, associated valves, and ascending aorta. Studies have shown intrinsic myocardial defects but do not sufficiently explain developmental defects in the endocardial-derived cardiac valve, septum, and vasculature. Here, we identify a developmentally impaired endocardial population in HLHS through single-cell RNA profiling of hiPSC-derived endocardium and human fetal heart tissue with an underdeveloped left ventricle. Intrinsic endocardial defects contribute to abnormal endothelial-to-mesenchymal transition, NOTCH signaling, and extracellular matrix organization, key factors in valve formation. Endocardial abnormalities cause reduced cardiomyocyte proliferation and maturation by disrupting fibronectin-integrin signaling, consistent with recently described de novo HLHS mutations associated with abnormal endocardial gene and fibronectin regulation. Together, these results reveal a critical role for endocardium in HLHS etiology and provide a rationale for considering endocardial function in regenerative strategies.
Keywords: ETS1; NOTCH; de novo mutation; endocardium; endothelial to mesenchymal transition; fibronectin; human heart tissue; hypoplastic left heart syndrome; induced pluripotent stem cells; single-cell RNA-seq.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Botto LD, Correa A, and Erickson JD (2001). Racial and temporal variations in the prevalence of heart defects. Pediatrics 107, E32. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
